

Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy
- PMID: 17186466
- PMCID: PMC1698714
- DOI: 10.1086/509044
Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy
Abstract
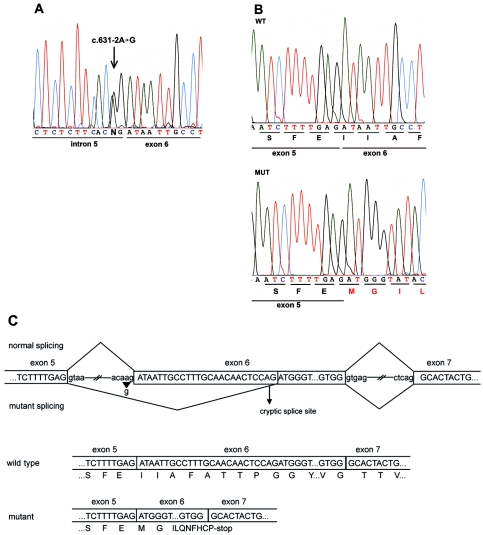
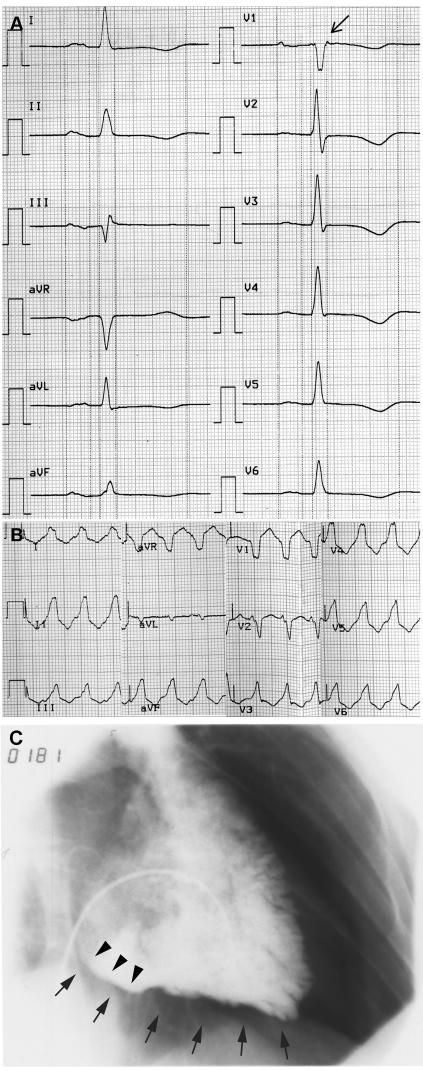
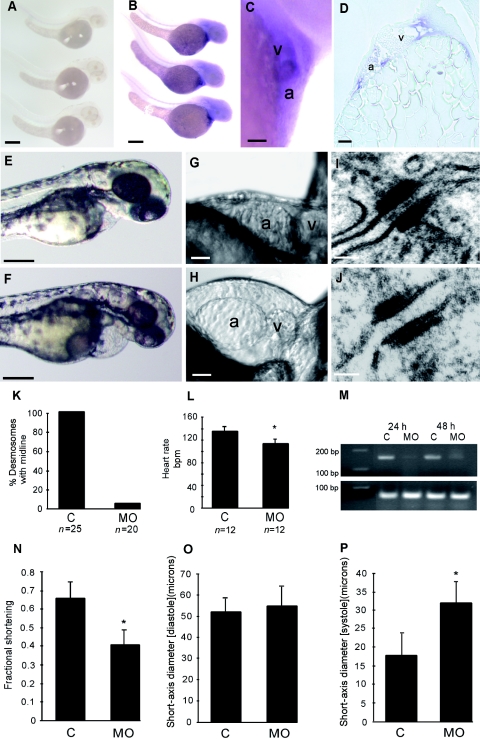
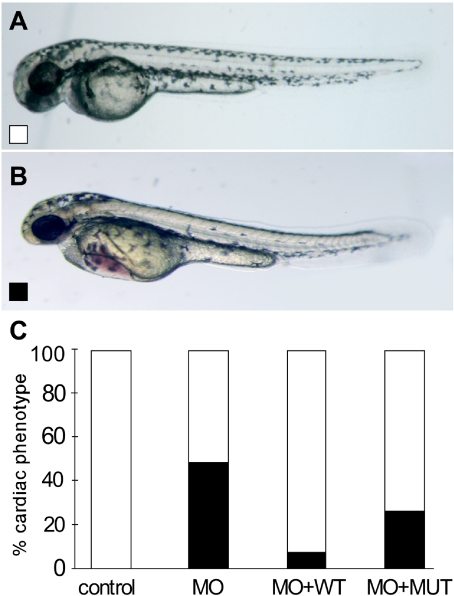
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetically heterogeneous heart-muscle disorder characterized by progressive fibrofatty replacement of right ventricular myocardium and an increased risk of sudden cardiac death. Mutations in desmosomal proteins that cause ARVC have been previously described; therefore, we investigated 88 unrelated patients with the disorder for mutations in human desmosomal cadherin desmocollin-2 (DSC2). We identified a heterozygous splice-acceptor-site mutation in intron 5 (c.631-2A-->G) of the DSC2 gene, which led to the use of a cryptic splice-acceptor site and the creation of a downstream premature termination codon. Quantitative analysis of cardiac DSC2 expression in patient specimens revealed a marked reduction in the abundance of the mutant transcript. Morpholino knockdown in zebrafish embryos revealed a requirement for dsc2 in the establishment of the normal myocardial structure and function, with reduced desmosomal plaque area, loss of the desmosome extracellular electron-dense midlines, and associated myocardial contractility defects. These data identify DSC2 mutations as a cause of ARVC in humans and demonstrate that physiologic levels of DSC2 are crucial for normal cardiac desmosome formation, early cardiac morphogenesis, and cardiac function.
Figures

References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for DSC2 transcript variant [accession number NM_024422.2] and zebrafish DNA from clone [accession number BX649302])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ARVC/D 1–10, DSC2, DSG2, PPKS1, and LAH)
References
-
- Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, Grosgogeat Y (1982) Right ventricular dysplasia: a report of 24 adult cases. Circulation 65:384–398 - PubMed
-
- Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988) Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 318:129–133 - PubMed
-
- Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Martini B, Stritoni P, Fasoli G (1988) Familial occurrence of right ventricular dysplasia: a study involving nine families. J Am Coll Cardiol 12:1222–1228 - PubMed
-
- Wlodarska EK, Konka M, Kepski R, Zaleska T, Ploski R, Ruzyllo W, Janion M, Jaworska K, Rydlewska-Sadowska W, Hoffman P (2004) Familial form of arrhythmogenic right ventricular cardiomyopathy. Kardiol Pol 60:1–14 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
