

Review
doi: 10.1016/j.bbamem.2006.10.021.
Epub 2006 Nov 15.
G protein coupled receptor structure and activation
Affiliations
- PMID: 17188232
- PMCID: PMC1876727
- DOI: 10.1016/j.bbamem.2006.10.021
Item in Clipboard
Review
G protein coupled receptor structure and activation
Biochim Biophys Acta.
2007 Apr.
Abstract
G protein coupled receptors (GPCRs) are remarkably versatile signaling molecules. The members of this large family of membrane proteins are activated by a spectrum of structurally diverse ligands, and have been shown to modulate the activity of different signaling pathways in a ligand specific manner. In this manuscript I will review what is known about the structure and mechanism of activation of GPCRs focusing primarily on two model systems, rhodopsin and the beta(2) adrenoceptor.
Figures

Cartoons depicting the secondary structure and the location of agonist binding sites for different GPCRs.
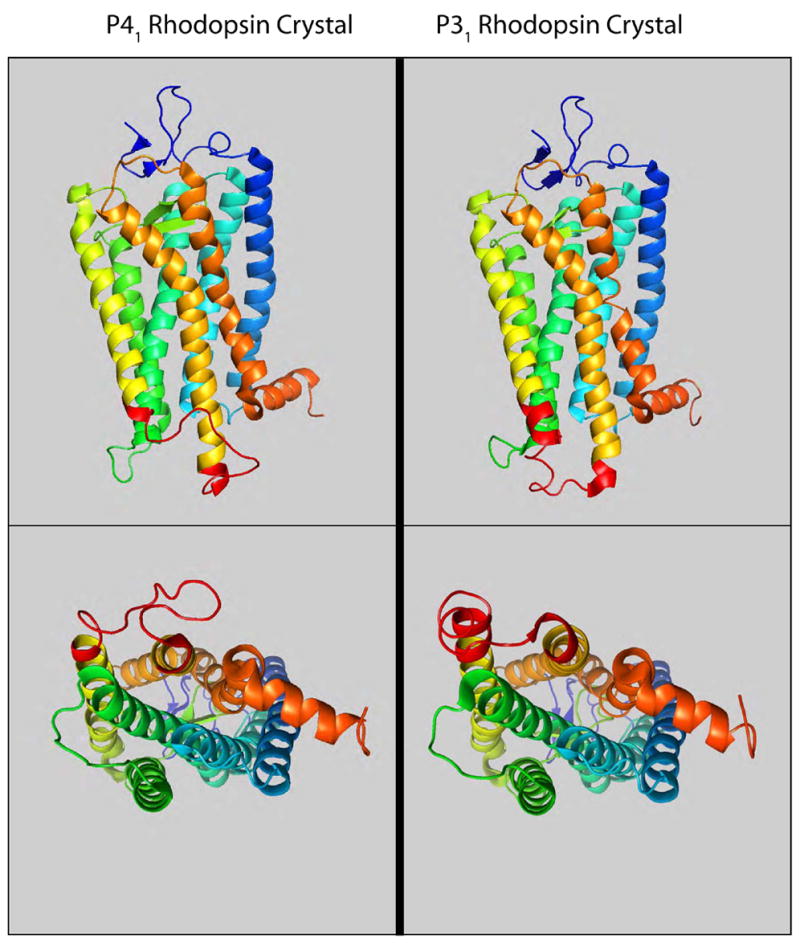
A comparison of the rhodopsin structures determined from the P41 [25] and P31 [26] crystal forms. The loop connecting TM5 and TM6 (shown in red) is the most divergent sequence.

Agonist binding site in the β2AR. A. Amino acids involved in forming the agonist binding site for the β2AR. The sites of interaction between catecholamines and the β2AR have been extensively characterized [–102]. The amine nitrogen interacts with Asp 113 in TM3 [103], the catechol hydroxyls interact with serines in TM5 [–102]. Interactions with the aromatic ring and the chiral β-hydroxyl have both been mapped to TM6 [101]. B. A panel of β2AR ligands discussed in this review. Catechol is a very weak partial agonist. Dopamine and salbutamol are partial agonists. Norepinephrine, epinephrine and isoproterenol are full agonists. ICI118,551 is an inverse agonist. D–E. Isoproterenol docked into a three-dimensional model of the β2AR. Illustrations were made with MacPyMOL software.
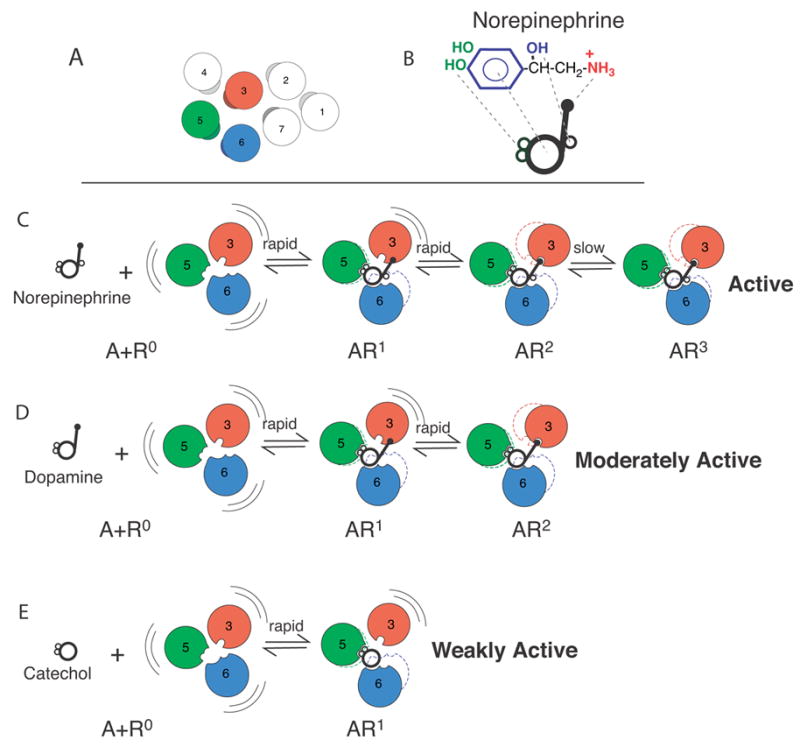
Sequential Binding Model. A. Arrangement of the TM domains of the β2AR as viewed from the extracellular surface. The agonist binding domains are shown in red (TM3), green (TM5) and blue (TM6). B. Cartoon representing structural components of norepinephrine. C–E. In the absence of ligand, the receptor (R) is conformationally flexible. Conformational state R1 is stabilized by interactions between TMs 5 and 6 and the catechol ring. The transition to state R2 occurs when Asp 113 in TM3 binds the amine nitrogen. The transitions from R to R2 are rapid. The slow transition from R2 to R3 involves interactions between the chiral β-hydroxyl and Asn293 on TM6. Adapted from Swaminath et al. [88].

Agonist-induced conformational changes detected by fluorescence spectroscopy in β2AR labeled at Cys265 with tetramethylrhodamine maleimide (TMR-β2AR). A. Change in the intensity of TMR-β2AR in response to norepinephrine and dopamine. The response to norepinephrine is best fit by a two-site exponential function. The rapid and slow components of the response are illustrated by the dotted lines. B. Change in the intensity of TMR-β2AR in response to dopamine and catechol. C. Catechol and dopamine stimulated [35S]GTPγS binding to purified β2AR reconstituted with purified Gs. D. The change in the intensity of TMR-β2AR in response to the non-catechol partial agonist salbutamol followed by the addition of catechol. Catechol can induce a conformational change in TMR-β2AR bound to a saturating concentration of salbutamol, indicating that salbutamol and catechol occupy non-overlapping binding sites. E. The change in the intensity of TMR-β2AR in response to norepinephrine followed by the addition of catechol. No catechol response is observed in TMR-β2AR bound to a saturating concentration of norepinephrine indicating that these ligands share a common binding site. F. There is no significant change in the intensity of TMR-β2AR in response to the inverse agonist ICI118,551. Catechol can induce a conformational change in β2AR bound to a saturating concentration of ICI118,551, indicating that these ligands do not occupy the same binding space. G. [35S]GTPγS binding to purified β2AR reconstituted with purified Gs. Catechol weakly stimulates [35S]GTPγS binding and ICI118,551 inhibits basal [35S]GTPγS binding. Of interest, catechol can stimulate [35S]GTPγS binding in β2AR occupied by a saturating concentration of ICI118,551. The data presented here are adapted from Swaminath et al. [88, 89].
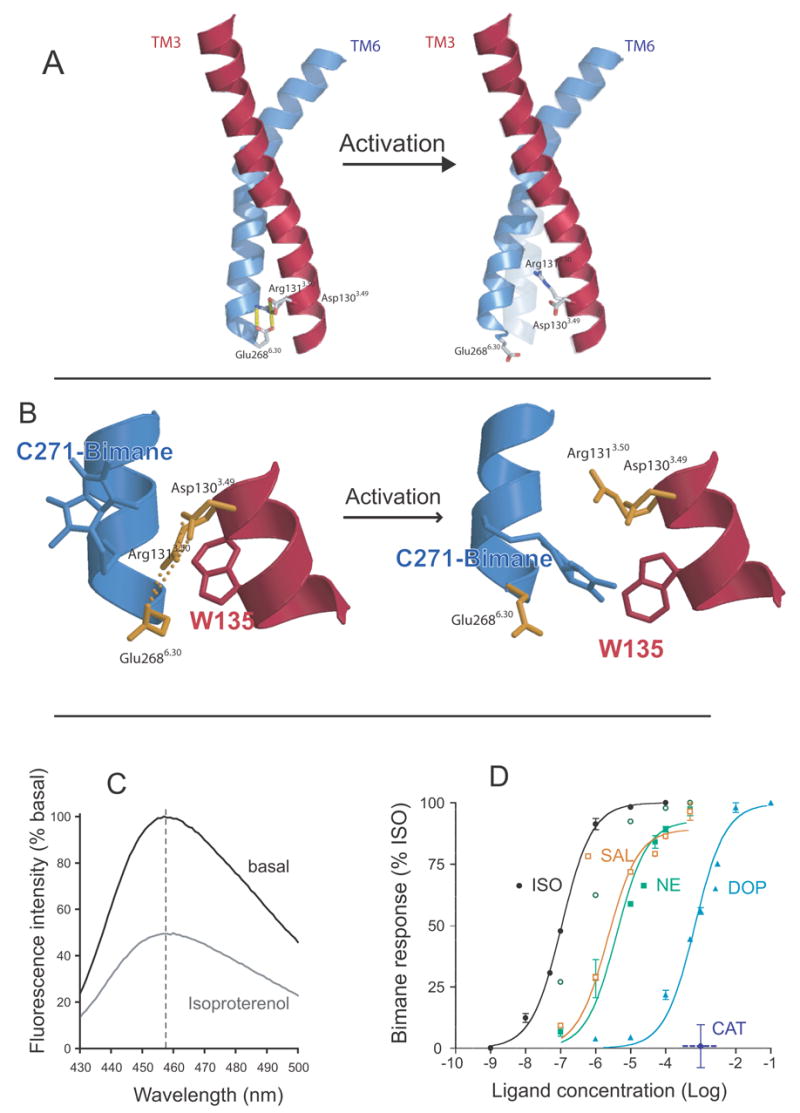
Fluorescence spectroscopy to monitor disruption of the ionic lock in the β2AR. A. Model of TM3 (red) and TM6 (blue) from the β2AR depicting the amino acids that comprise the ionic lock at the cytoplasmic end of these TM segments. B. Close up view of the ionic lock and the modifications made to monitor conformational changes in this region. Alanine 271 was mutated to cysteine (C271) and isoleucine 135 was mutated to tryptophan (W135). C271 was labeled with monobromobimane in purified β2AR. Upon activation, W135 moves closer to bimane on C271 and quenches fluorescence. C. Emission spectrum of bimane on C271 before and after activation by the agonist isoproterenol. D. Effect of different ligands on disruption of the ionic lock as determined by bimane fluorescence. The partial agonists dopamine and salbutamol are as effective at disrupting the ionic lock as the full agonists norepinephrine and isoproterenol. Only catechol has no effect on the ionic lock. These data are adapted from Yao et al. [71].
References
-
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein coupled receptors. Meth Neurosci. 1995;25:366–428.
-
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308(5721):512–7. - PubMed
-
- Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115(Pt 3):455–65. - PubMed
-
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
