

Anatomy of Escherichia coli sigma70 promoters
- PMID: 17189297
- PMCID: PMC1807945
- DOI: 10.1093/nar/gkl956
Anatomy of Escherichia coli sigma70 promoters
Abstract
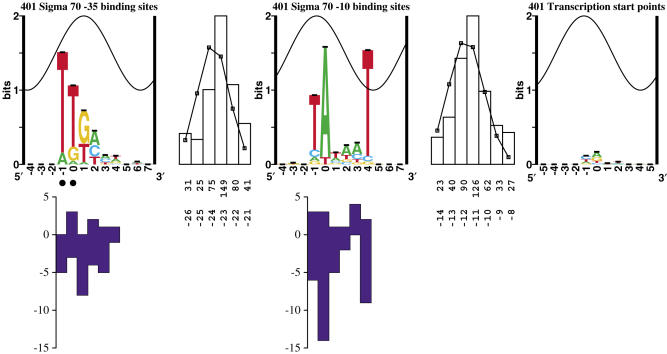
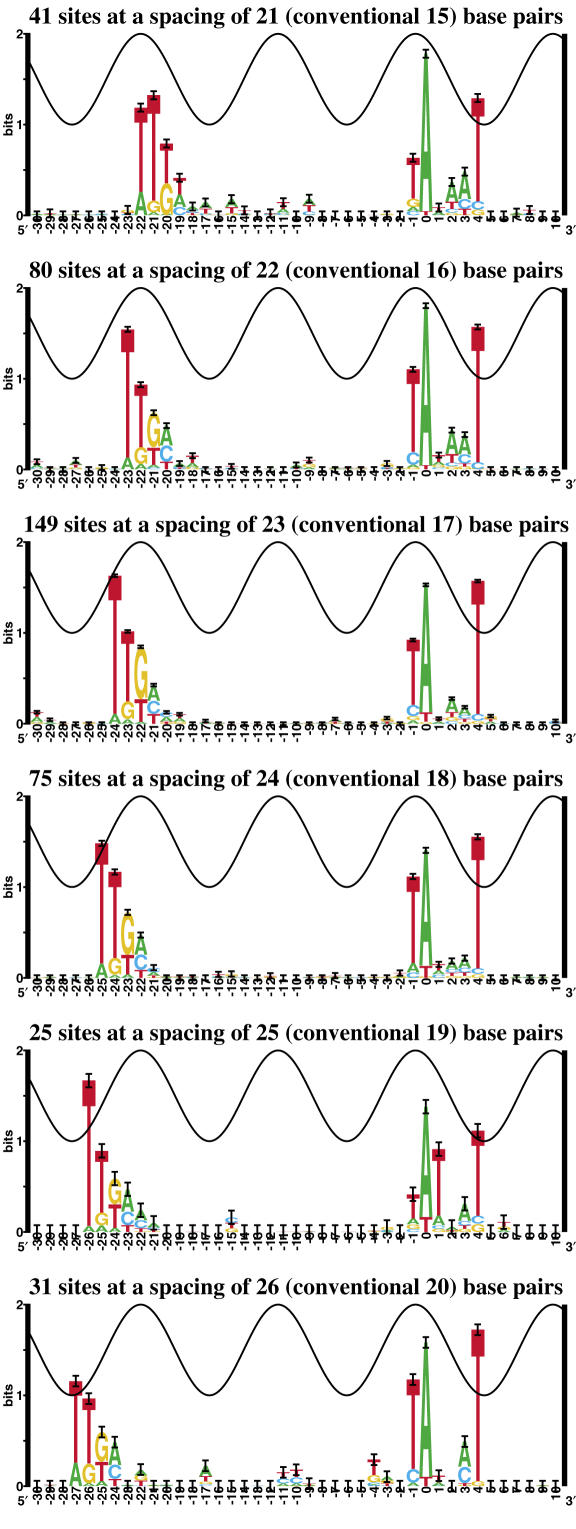
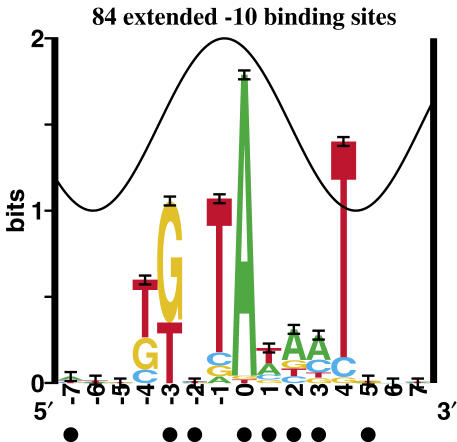
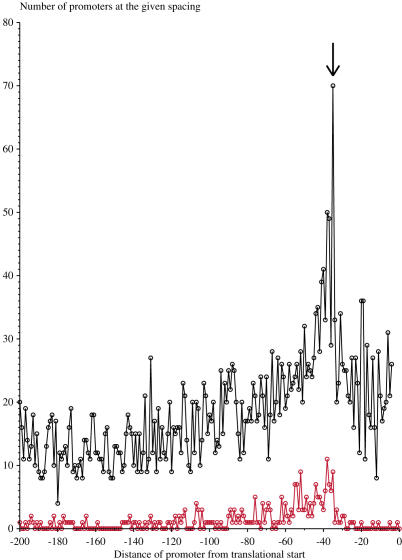
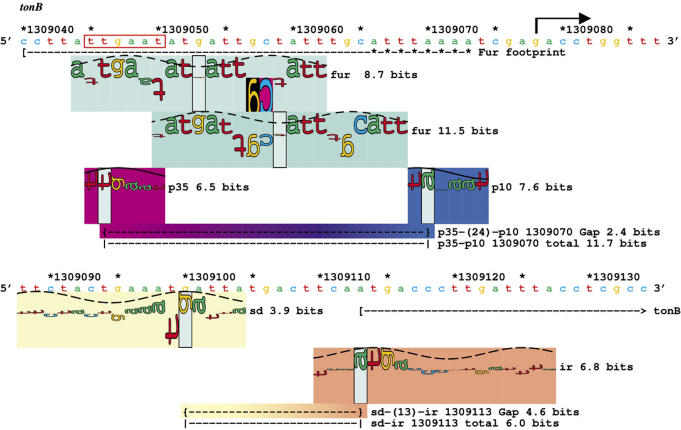
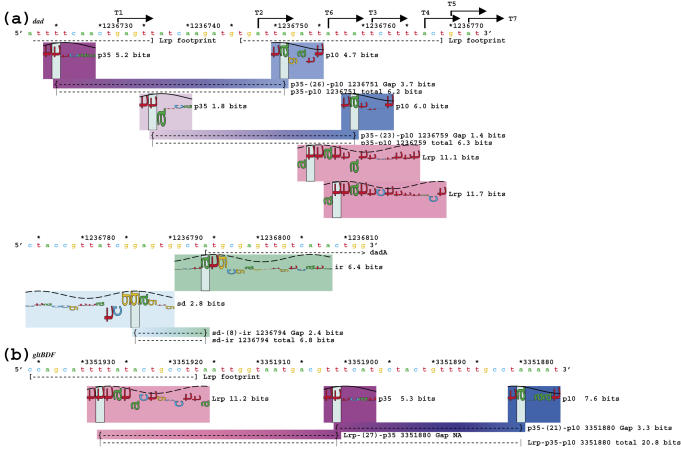
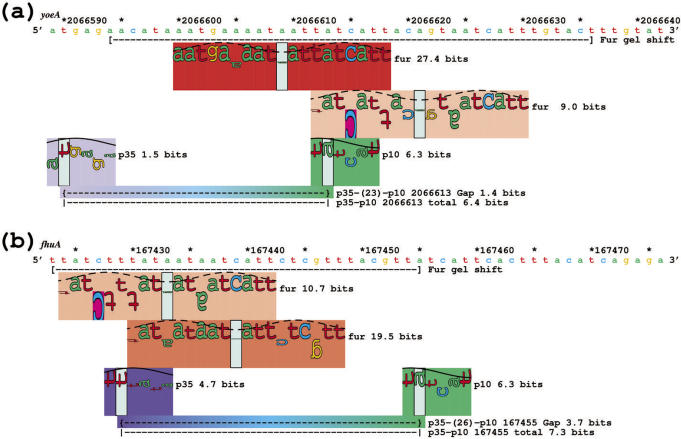
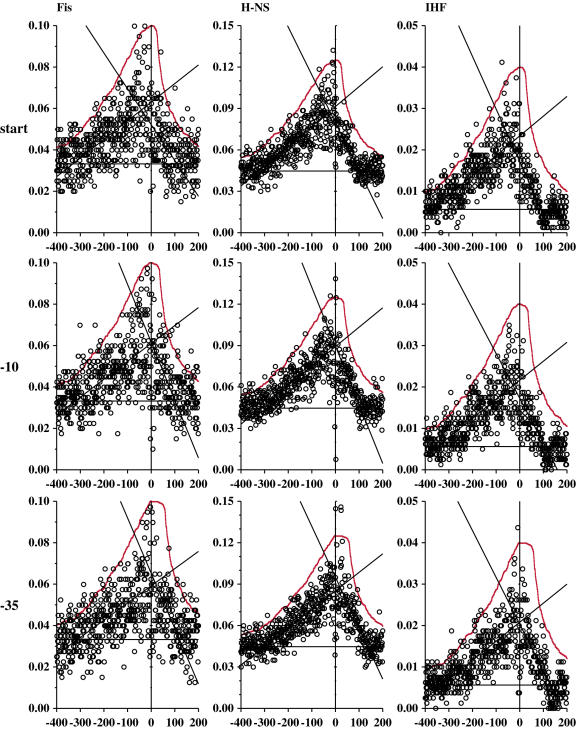
Information theory was used to build a promoter model that accounts for the -10, the -35 and the uncertainty of the gap between them on a common scale. Helical face assignment indicated that base -7, rather than -11, of the -10 may be flipping to initiate transcription. We found that the sequence conservation of sigma70 binding sites is 6.5 +/- 0.1 bits. Some promoters lack a -35 region, but have a 6.7 +/- 0.2 bit extended -10, almost the same information as the bipartite promoter. These results and similarities between the contacts in the extended -10 binding and the -35 suggest that the flexible bipartite sigma factor evolved from a simpler polymerase. Binding predicted by the bipartite model is enriched around 35 bases upstream of the translational start. This distance is the smallest 5' mRNA leader necessary for ribosome binding, suggesting that selective pressure minimizes transcript length. The promoter model was combined with models of the transcription factors Fur and Lrp to locate new promoters, to quantify promoter strengths, and to predict activation and repression. Finally, the DNA-bending proteins Fis, H-NS and IHF frequently have sites within one DNA persistence length from the -35, so bending allows distal activators to reach the polymerase.
Figures
References
-
- Horwitz M.S., Loeb L.A. Structure–function relationships in Escherichia coli promoter DNA. Prog. Nucleic Acid Res. Mol. Biol. 1990;38:137–164. - PubMed
-
- Gralla J.D. Promoter recognition and mRNA initiation by Escherichia coli E σ70. Methods Enzymol. 1990;185:37–54. - PubMed
-
- Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. - PubMed
-
- Burgess R.R., Travers A.A., Dunn J.J., Bautz E.K. Factor stimulating transcription by RNA polymerase. Nature. 1969;221:43–46. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
