

dSLAM analysis of genome-wide genetic interactions in Saccharomyces cerevisiae
- PMID: 17189863
- PMCID: PMC2491713
- DOI: 10.1016/j.ymeth.2006.07.033
dSLAM analysis of genome-wide genetic interactions in Saccharomyces cerevisiae
Abstract
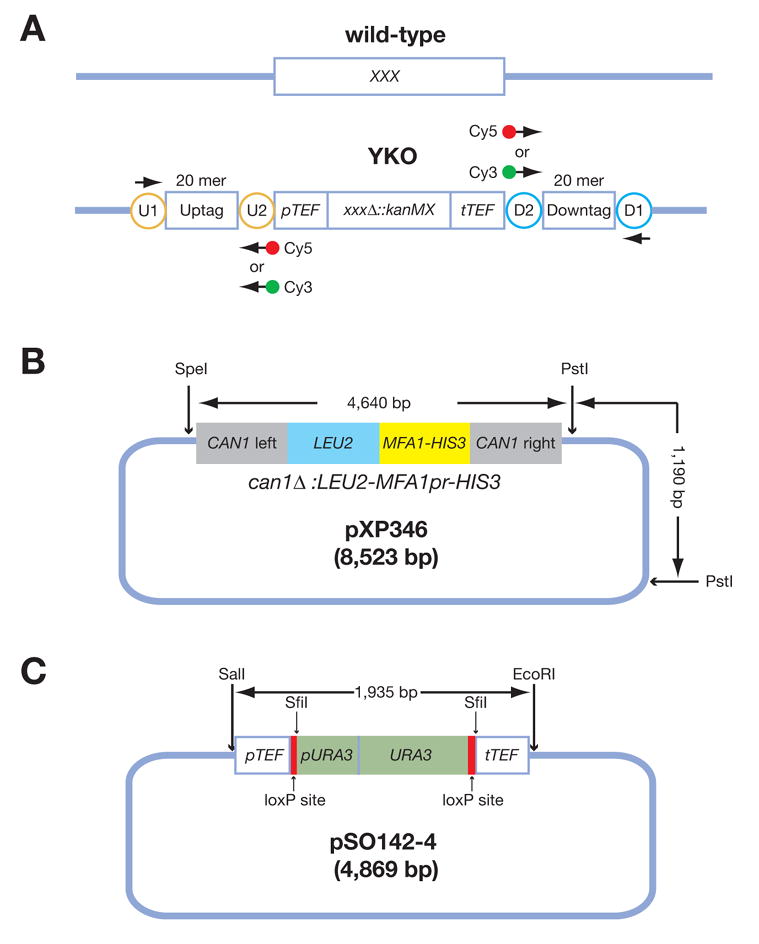
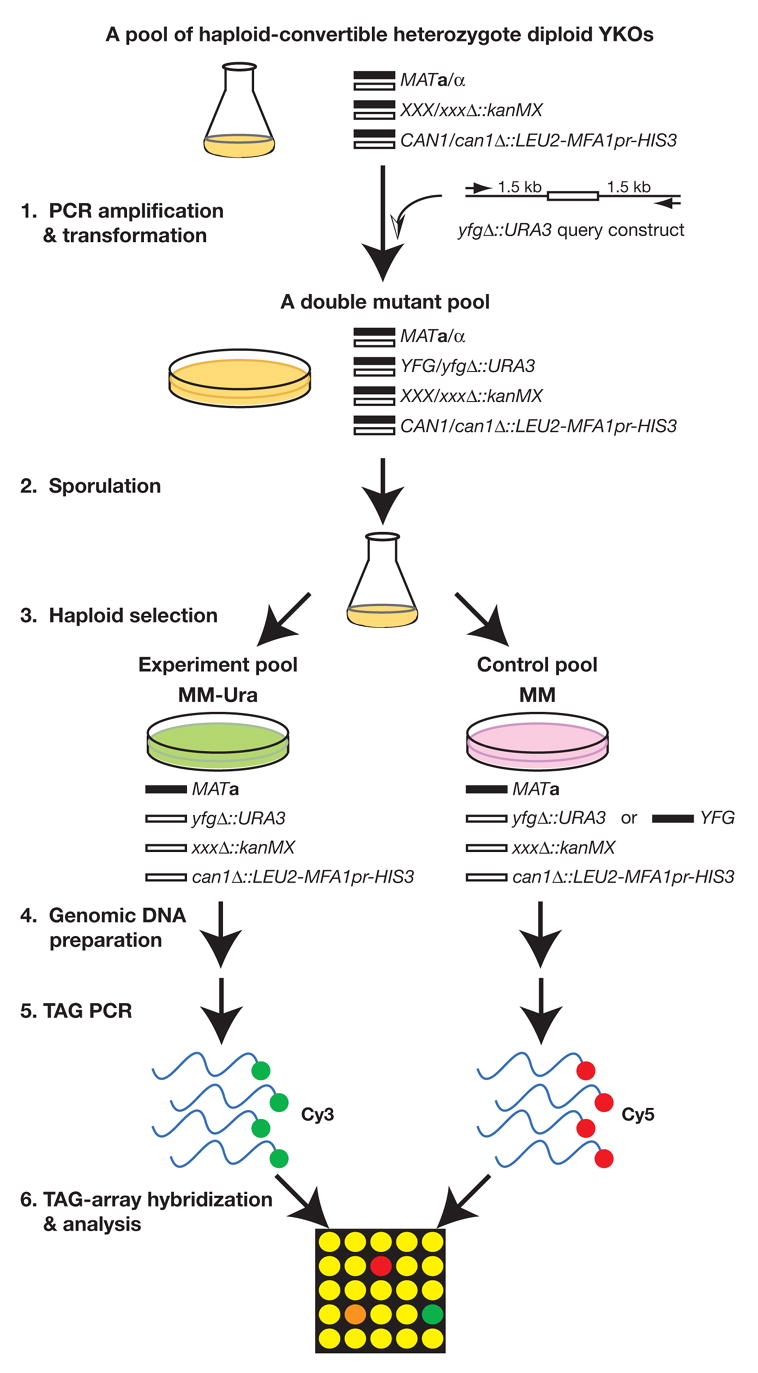
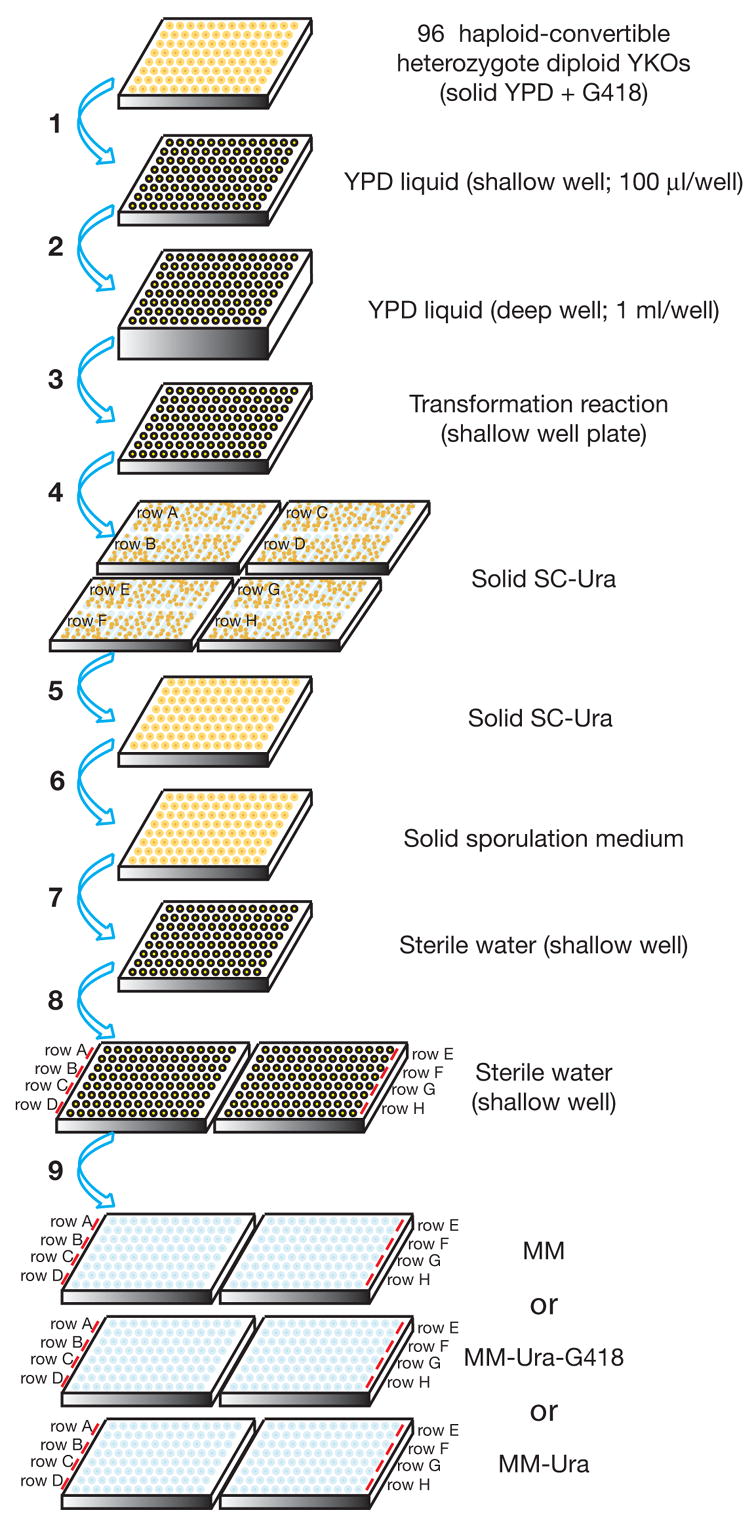
Analysis of genetic interactions has been extensively exploited to study gene functions and to dissect pathway structures. One such genetic interaction is synthetic lethality, in which the combination of two non-lethal mutations leads to loss of organism viability. We have developed a dSLAM (heterozygote diploid-based synthetic lethality analysis with microarrays) technology that effectively studies synthetic lethality interactions on a genome-wide scale in the budding yeast Saccharomyces cerevisiae. Typically, a query mutation is introduced en masse into a population of approximately 6000 haploid-convertible heterozygote diploid Yeast Knockout (YKO) mutants via integrative transformation. Haploid pools of single and double mutants are freshly generated from the resultant heterozygote diploid double mutant pool after meiosis and haploid selection and studied for potential growth defects of each double mutant combination by microarray analysis of the "molecular barcodes" representing each YKO. This technology has been effectively adapted to study other types of genome-wide genetic interactions including gene-compound synthetic lethality, secondary mutation suppression, dosage-dependent synthetic lethality and suppression.
Figures
References
-
- Hartman JLt, Garvik B, Hartwell L. Science. 2001;291:1001–4. - PubMed
-
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Davis RW, et al. Science. 1999;285:901–6. - PubMed
-
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Nature. 2002;418:387–91. - PubMed
-
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ. Cell. 2005;123:507–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
