

DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse
- PMID: 17190840
- PMCID: PMC2118439
- DOI: 10.1084/jem.20062131
DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse
Abstract
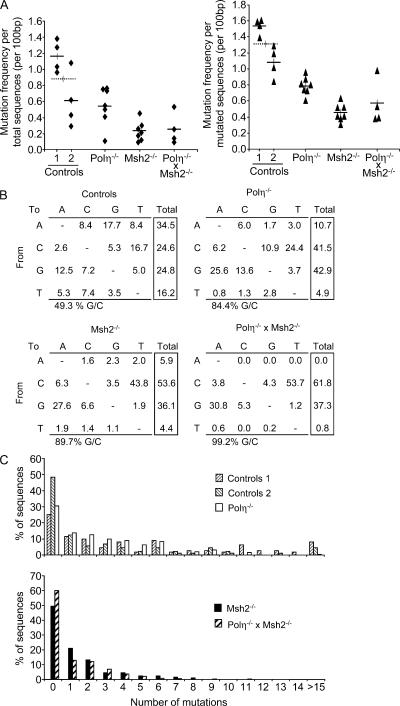
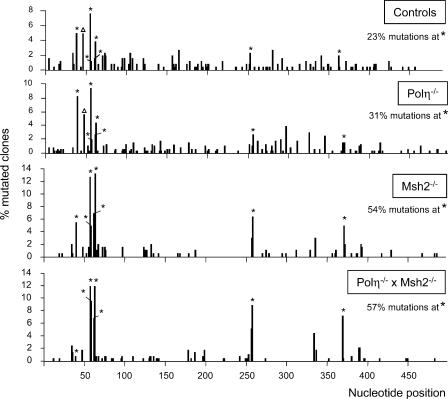
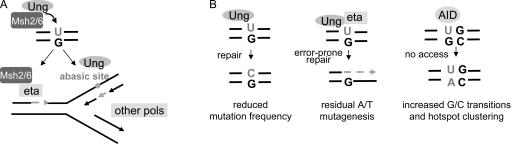
Mutations at A/T bases within immunoglobulin genes have been shown to be generated by a repair pathway involving the DNA-binding moiety of the mismatch repair complex constituted by the MSH2-MSH6 proteins, together with DNA polymerase eta (pol eta). However, residual A/T mutagenesis is still observed upon inactivation in the mouse of each of these factors, suggesting that the panel of activities involved might be more complex. We reported previously (Delbos, F., A. De Smet, A. Faili, S. Aoufouchi, J.-C. Weill, and C.-A. Reynaud. 2005. J. Exp. Med. 201:1191-1196) that residual A/T mutagenesis in pol eta-deficient mice was likely contributed by another enzyme not normally involved in hypermutation, DNA polymerase kappa, which is mobilized in the absence of the normal polymerase partner. We report the complete absence of A/T mutations in MSH2-pol eta double-deficient mice, thus indicating that the residual A/T mutagenesis in MSH2-deficient mice is contributed by pol eta, now recruited by uracil N-glycosylase, the second DNA repair pathway involved in hypermutation. We propose that this particular recruitment of pol eta corresponds to a profound modification of the function of uracil glycosylase in the absence of the mismatch repair complex, suggesting that MSH2-MSH6 actively prevent uracil glycosylase from error-free repair during hypermutation. pol eta thus appears to be the sole contributor of A/T mutations in the normal physiological context.
Figures
References
-
- Longo, N.S., and P.E. Lipsky. 2006. Why do B cells mutate their immunoglobulin receptors? Trends Immunol. 27:374–380. - PubMed
-
- Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. - PubMed
-
- Longerich, S., U. Basu, F.W. Alt, and U. Storb. 2006. AID in somatic hypermutation and class switch recombination. Curr. Opin. Immunol. 18:164–174. - PubMed
-
- Odegard, V.H., and D.G. Schatz. 2006. Targeting of somatic hypermutation. Nat. Rev. Immunol. 6:573–583. - PubMed
-
- Reynaud, C.-A., S. Aoufouchi, A. Faili, and J.-C. Weill. 2003. What role for AID: mutator, or assembler of the immunoglobulin mutasome? Nat. Immunol. 4:631–638. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
