

Connectivity in the yeast cell cycle transcription network: inferences from neural networks
- PMID: 17194216
- PMCID: PMC1761652
- DOI: 10.1371/journal.pcbi.0020169
Connectivity in the yeast cell cycle transcription network: inferences from neural networks
Abstract
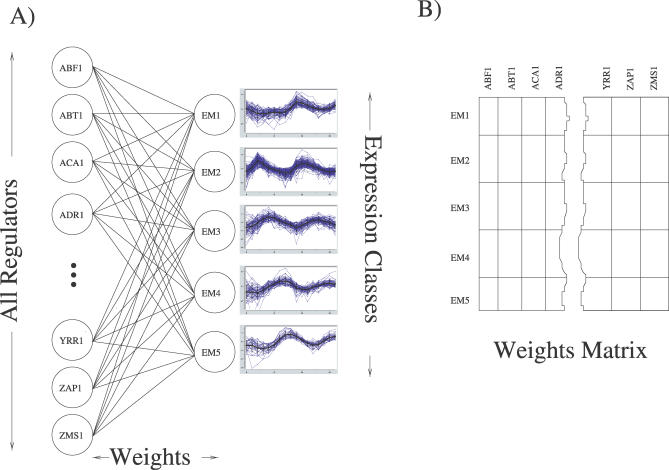
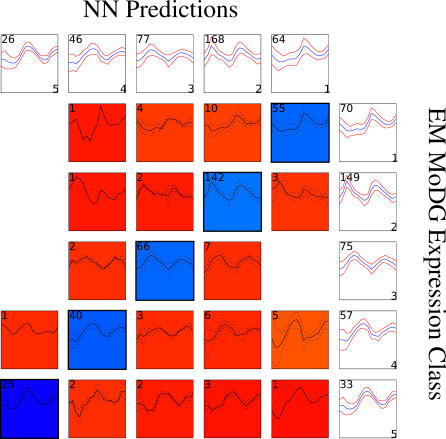
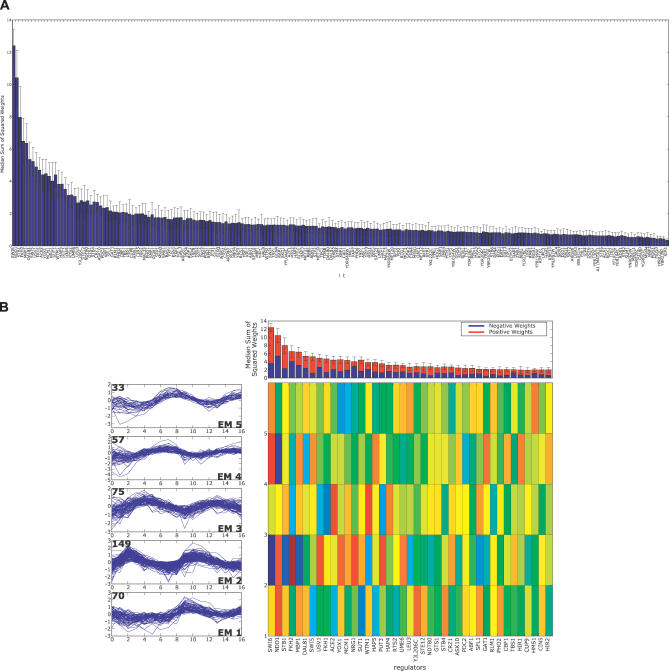
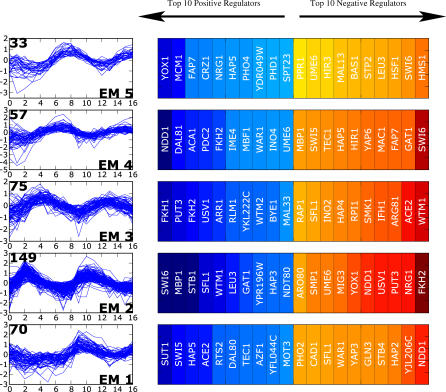
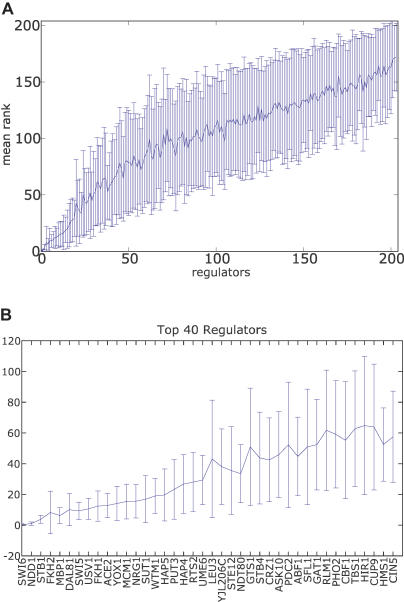
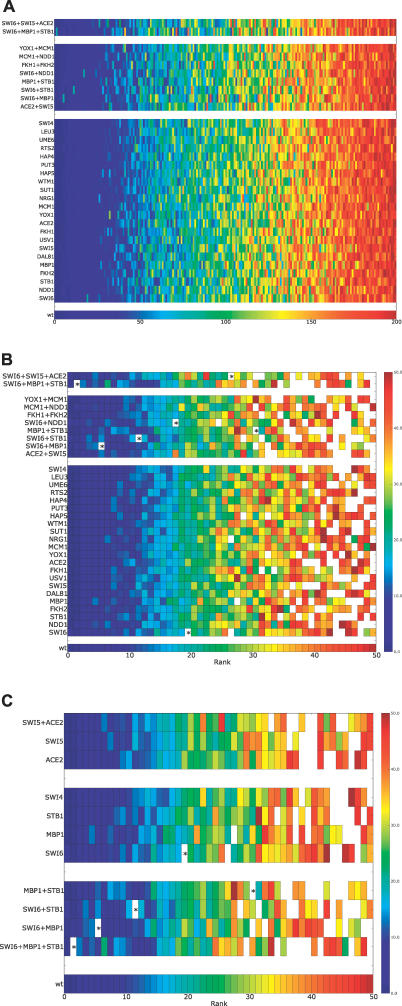
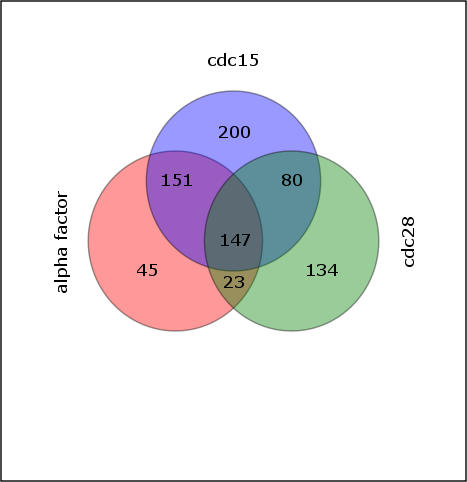
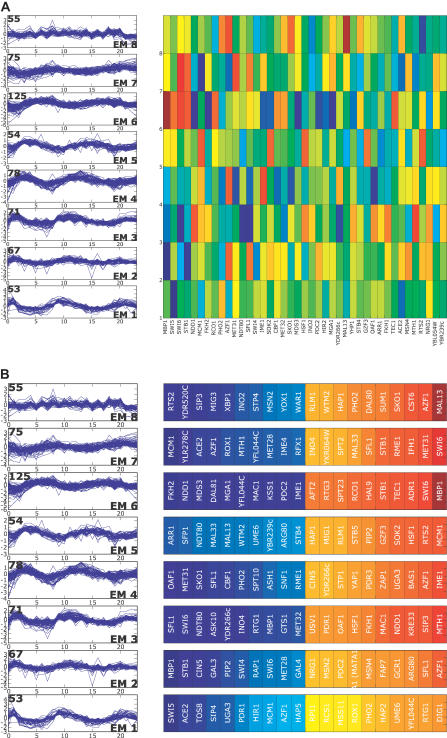
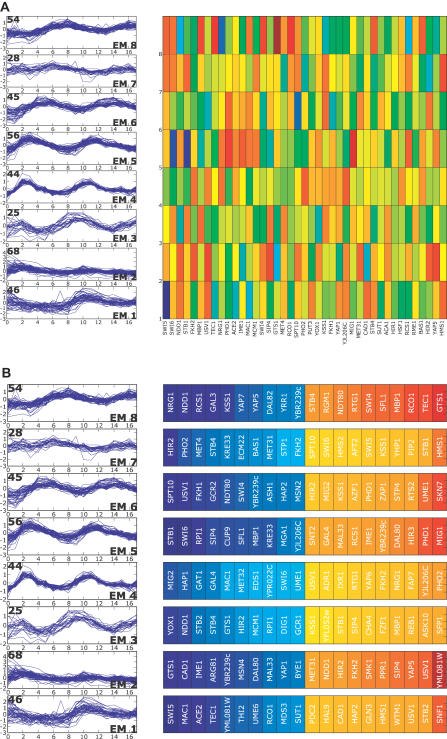
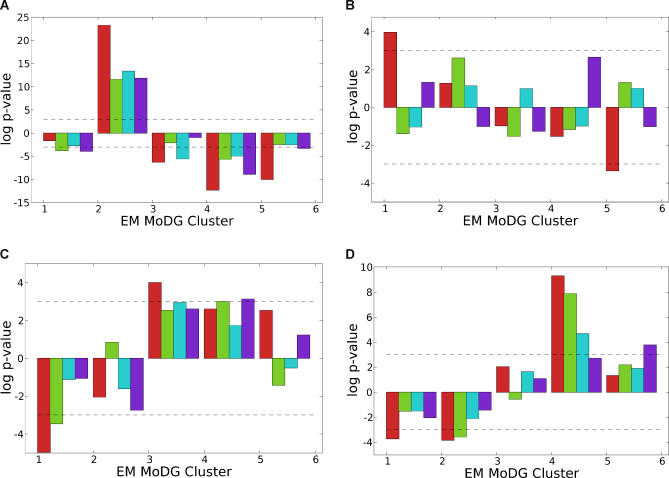
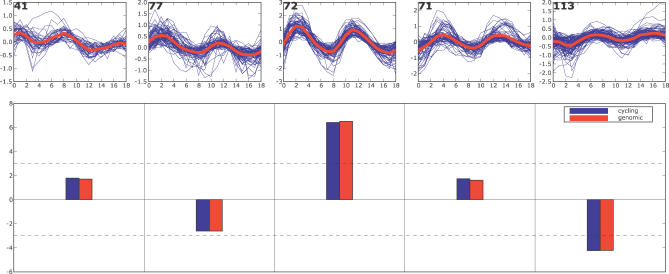
A current challenge is to develop computational approaches to infer gene network regulatory relationships based on multiple types of large-scale functional genomic data. We find that single-layer feed-forward artificial neural network (ANN) models can effectively discover gene network structure by integrating global in vivo protein:DNA interaction data (ChIP/Array) with genome-wide microarray RNA data. We test this on the yeast cell cycle transcription network, which is composed of several hundred genes with phase-specific RNA outputs. These ANNs were robust to noise in data and to a variety of perturbations. They reliably identified and ranked 10 of 12 known major cell cycle factors at the top of a set of 204, based on a sum-of-squared weights metric. Comparative analysis of motif occurrences among multiple yeast species independently confirmed relationships inferred from ANN weights analysis. ANN models can capitalize on properties of biological gene networks that other kinds of models do not. ANNs naturally take advantage of patterns of absence, as well as presence, of factor binding associated with specific expression output; they are easily subjected to in silico "mutation" to uncover biological redundancies; and they can use the full range of factor binding values. A prominent feature of cell cycle ANNs suggested an analogous property might exist in the biological network. This postulated that "network-local discrimination" occurs when regulatory connections (here between MBF and target genes) are explicitly disfavored in one network module (G2), relative to others and to the class of genes outside the mitotic network. If correct, this predicts that MBF motifs will be significantly depleted from the discriminated class and that the discrimination will persist through evolution. Analysis of distantly related Schizosaccharomyces pombe confirmed this, suggesting that network-local discrimination is real and complements well-known enrichment of MBF sites in G1 class genes.
Conflict of interest statement
Figures
Similar articles
-
Transcriptome network component analysis with limited microarray data.Bioinformatics. 2006 Aug 1;22(15):1886-94. doi: 10.1093/bioinformatics/btl279. Epub 2006 Jun 9. Bioinformatics. 2006. PMID: 16766556
-
A Gibbs sampler for the identification of gene expression and network connectivity consistency.Bioinformatics. 2006 Dec 15;22(24):3040-6. doi: 10.1093/bioinformatics/btl541. Epub 2006 Oct 23. Bioinformatics. 2006. PMID: 17060361
-
A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data.Bioinformatics. 2005 Jan 1;21(1):71-9. doi: 10.1093/bioinformatics/bth463. Epub 2004 Aug 12. Bioinformatics. 2005. PMID: 15308537
-
Inferring network interactions within a cell.Brief Bioinform. 2005 Dec;6(4):380-9. doi: 10.1093/bib/6.4.380. Brief Bioinform. 2005. PMID: 16420736 Review.
-
Artificial intelligence in hematology.Hematology. 2005 Oct;10(5):393-400. doi: 10.1080/10245330410001727055. Hematology. 2005. PMID: 16203606 Review.
Cited by
-
Context-dependent DNA recognition code for C2H2 zinc-finger transcription factors.Bioinformatics. 2008 Sep 1;24(17):1850-7. doi: 10.1093/bioinformatics/btn331. Epub 2008 Jun 27. Bioinformatics. 2008. PMID: 18586699 Free PMC article.
-
Systems biology of interstitial lung diseases: integration of mRNA and microRNA expression changes.BMC Med Genomics. 2011 Jan 17;4:8. doi: 10.1186/1755-8794-4-8. BMC Med Genomics. 2011. PMID: 21241464 Free PMC article.
-
Stochastic spatio-temporal dynamic model for gene/protein interaction network in early Drosophila development.Gene Regul Syst Bio. 2009 Oct 19;3:191-210. Gene Regul Syst Bio. 2009. PMID: 20054403 Free PMC article.
-
Modeling regulatory cascades using Artificial Neural Networks: the case of transcriptional regulatory networks shaped during the yeast stress response.Front Genet. 2013 Jun 20;4:110. doi: 10.3389/fgene.2013.00110. eCollection 2013. Front Genet. 2013. PMID: 23802010 Free PMC article.
-
Models from experiments: combinatorial drug perturbations of cancer cells.Mol Syst Biol. 2008;4:216. doi: 10.1038/msb.2008.53. Epub 2008 Sep 2. Mol Syst Biol. 2008. PMID: 18766176 Free PMC article.
References
-
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. - PubMed
-
- Rustici G, Mata J, Kivinen K, Lio P, Penkett CJ, et al. Periodic gene expression program of the fission yeast cell cycle. Nat Genet. 2004;8:809–817. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
