

Substitution of critical isoleucines in the KH domains of Drosophila fragile X protein results in partial loss-of-function phenotypes
- PMID: 17194772
- PMCID: PMC1840061
- DOI: 10.1534/genetics.106.068908
Substitution of critical isoleucines in the KH domains of Drosophila fragile X protein results in partial loss-of-function phenotypes
Abstract
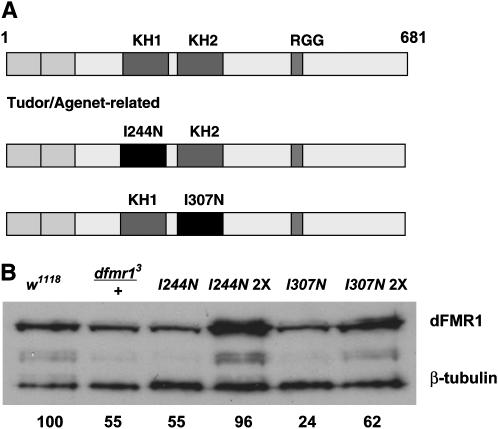
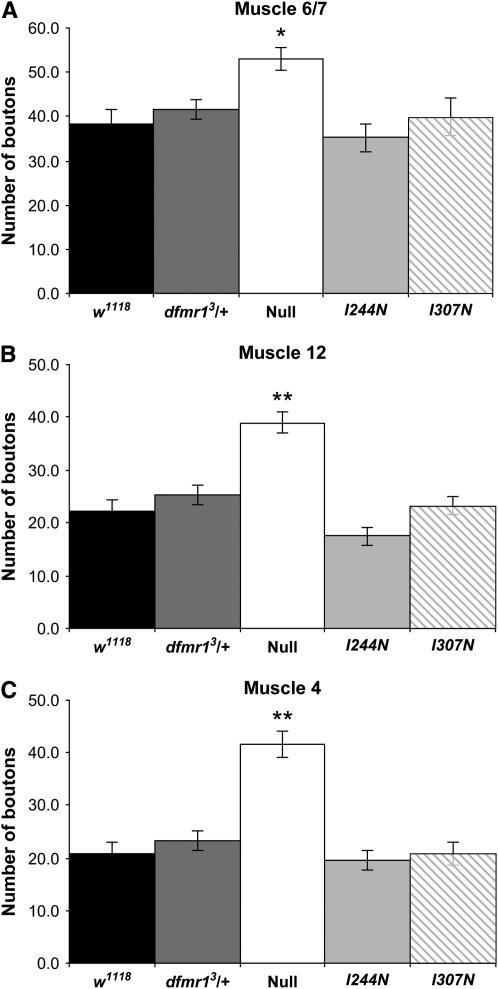
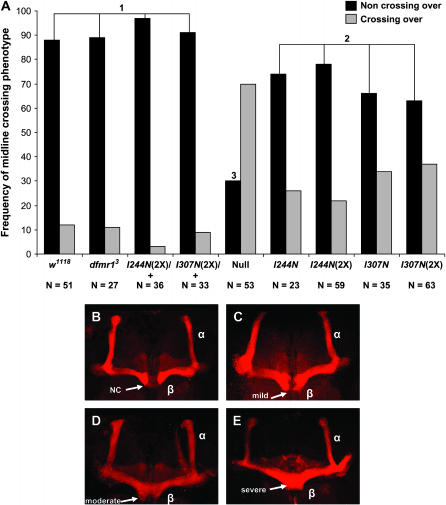
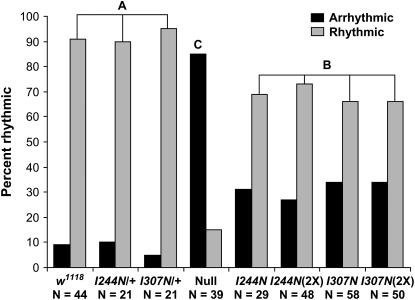
Fragile X mental retardation proteins (FMRP) are RNA-binding proteins that interact with a subset of cellular RNAs. Several RNA-binding domains have been identified in FMRP, but the contribution of these individual domains to FMRP function in an animal model is not well understood. In this study, we have generated flies with point mutations in the KH domains of the Drosophila melanogaster fragile X gene (dfmr1) in the context of a genomic rescue fragment. The substitutions of conserved isoleucine residues within the KH domains with asparagine are thought to impair binding of RNA substrates and perhaps the ability of FMRP to assemble into mRNP complexes. The mutants were analyzed for defects in development and behavior that are associated with deletion null alleles of dfmr1. We find that these KH domain mutations result in partial loss of function or no significant loss of function for the phenotypes assayed. The phenotypes resulting from these KH domain mutants imply that the capacities of the mutant proteins to bind RNA and form functional mRNP complexes are not wholly disrupted and are consistent with biochemical models suggesting that RNA-binding domains of FMRP can function independently.
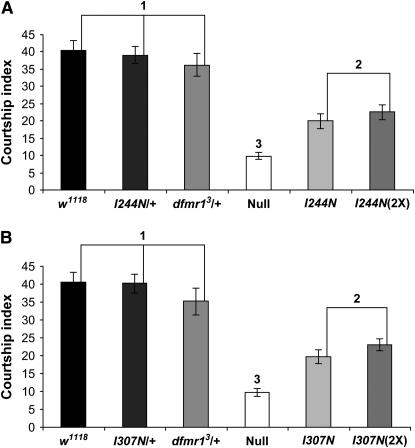
Figures
References
-
- Adinolfi, S., A. Ramos, S. R. Martin, F. Dal Piaz, P. Pucci et al., 2003. The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry 42: 10437–10444. - PubMed
-
- Ashley, C. T., Jr., K. D. Wilkinson, D. Reines and S. T. Warren, 1993. FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262: 563–566. - PubMed
-
- Bagni, C., and W. T. Greenough, 2005. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6: 376–387. - PubMed
-
- Bardoni, B., and J. L. Mandel, 2002. Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr. Opin. Genet. Dev. 12: 284–293. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
