

A sodium channel pore mutation causing Brugada syndrome
- PMID: 17198989
- PMCID: PMC1779366
- DOI: 10.1016/j.hrthm.2006.09.031
A sodium channel pore mutation causing Brugada syndrome
Abstract
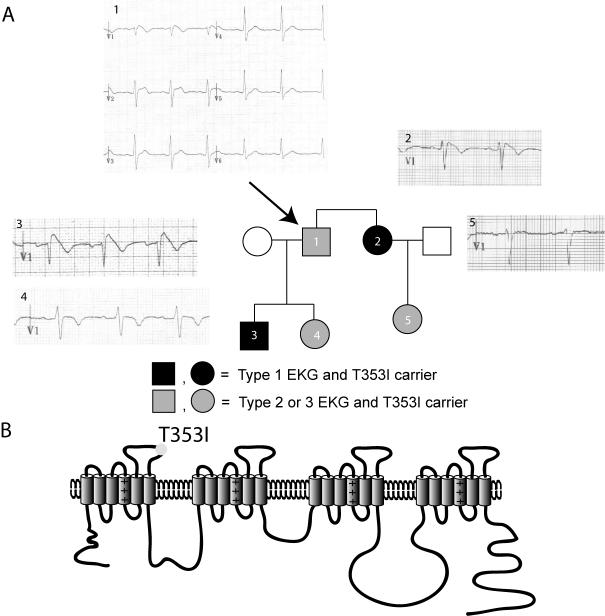
Background: Brugada and long QT type 3 syndromes are linked to sodium channel mutations and clinically cause arrhythmias that lead to sudden death. We have identified a novel threonine-to-isoleucine missense mutation at position 353 (T353I) adjacent to the pore-lining region of domain I of the cardiac sodium channel (SCN5A) in a family with Brugada syndrome. Both male and female carriers are symptomatic at young ages, have typical Brugada-type electrocardiogram changes, and have relatively normal corrected QT intervals.
Objectives: To characterize the properties of the newly identified cardiac sodium channel (SCN5A) mutation at the cellular level.
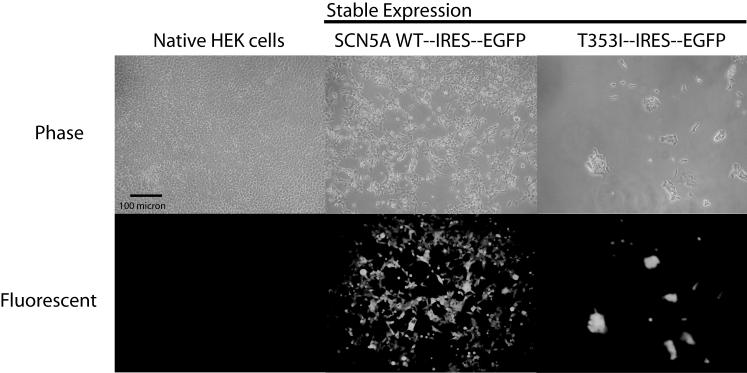
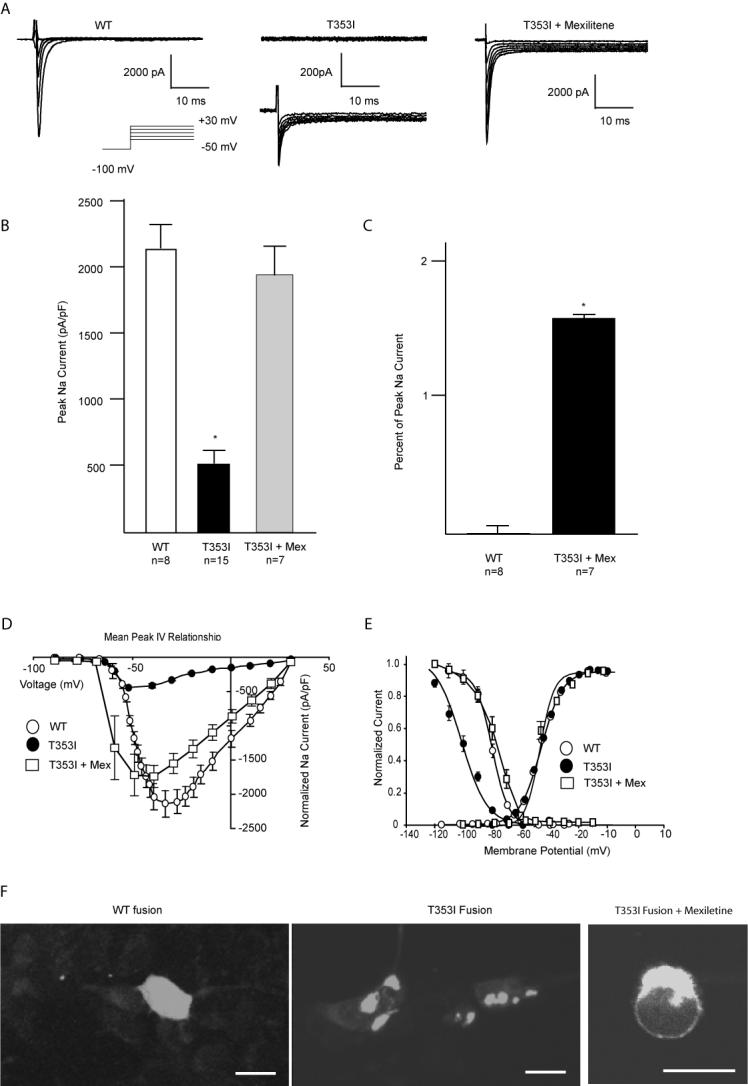
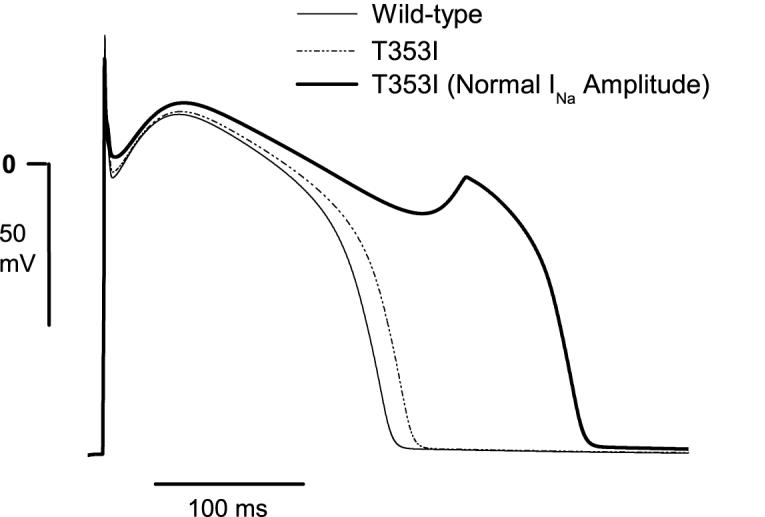
Results: Using whole-cell voltage clamp, we found that heterologous expression of SCN5A containing the T353I mutation resulted in 74% +/- 6% less peak macroscopic sodium current when compared with wild-type channels. A construct of the T353I mutant channel fused with green fluorescent protein failed to traffic properly to the sarcolemma, with a large proportion of channels sequestered intracellularly. Overnight exposure to 0.1 mM mexiletine, a Na(+) channel blocking agent, increased T353I channel trafficking to the membrane to near normal levels, but the mutant channels showed a significant late current that was 1.6% +/- 0.2% of peak sodium current at 200 ms, a finding seen with long QT mutations.
Conclusions: The clinical presentation of patients carrying the T353I mutation is that of Brugada syndrome and could be explained by a cardiac Na(+) channel trafficking defect. However, when the defect was ameliorated, the mutated channels had biophysical properties consistent with long QT syndrome. The lack of phenotypic changes associated with the long QT syndrome could be explained by a T353I-induced trafficking defect reducing the number of mutant channels with persistent currents present at the sarcolemma.
Figures
Comment in
-
Just another Brugada syndrome mutation?Heart Rhythm. 2007 Jan;4(1):54-5. doi: 10.1016/j.hrthm.2006.11.006. Epub 2006 Nov 17. Heart Rhythm. 2007. PMID: 17198990 No abstract available.
References
-
- Amin AS, Verkerk AO, Bhuiyan ZA, Wilde AA, Tan HL. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol Scand. 2005;185:291–301. - PubMed
-
- Baroudi G, Napolitano C, Priori SG, Del BA, Chahine M. Loss of function associated with novel mutations of the SCN5A gene in patients with Brugada syndrome. Can J Cardiol. 2004;20:425–30. - PubMed
-
- Baroudi G, Acharfi S, Larouche C, Chahine M. Expression and intracellular localization of an SCN5A double mutant R1232W/T1620M implicated in Brugada syndrome. Circ Res. 2002;90:E11–E16. - PubMed
-
- Baroudi G, Pouliot V, Denjoy I, Guicheney P, Shrier A, Chahine M. Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G) Circ Res. 2001;88:E78–E83. - PubMed
-
- Vatta M, Dumaine R, Antzelevitch C, Brugada R, Li H, Bowles NE, Nademanee K, Brugada J, Brugada P, Towbin JA. Novel mutations in domain I of SCN5A cause Brugada syndrome. Mol Genet and Metab. 2002;75:317–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
