

Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies
- PMID: 17200182
- PMCID: PMC1762697
- DOI: 10.2353/ajpath.2007.060573
Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies
Abstract
Clinical and experimental data indicate that anti-neutrophil cytoplasmic autoantibodies (ANCAs) cause glomerulonephritis and vasculitis. Here we report the first evidence that complement is an important mediator of ANCA disease. Transfer of anti-myeloperoxidase (MPO) IgG into wild-type mice or anti-MPO splenocytes into immune-deficient mice caused crescentic glomerulonephritis that could be completely blocked by complement depletion. The role of specific complement activation pathways was investigated using mice with knockout of the common pathway component C5, classic and lectin binding pathway component C4, and alternative pathway component factor B. After injection of anti-MPO IgG, C4-/- mice developed disease comparable with wild-type disease; however, C5-/- and factor B-/- mice developed no disease. To substantiate a role for complement in human ANCA disease, IgG was isolated from patients with myeloperoxidase ANCA (MPO-ANCA) or proteinase 3 ANCA (PR3-ANCA) and from controls. Incubation of MPO-ANCA or PR3-ANCA IgG with human neutrophils caused release of factors that activated complement. IgG from healthy controls did not produce this effect. The findings suggest that stimulation of neutrophils by ANCA causes release of factors that activate complement via the alternative pathway, thus initiating an inflammatory amplification loop that mediates the severe necrotizing inflammation of ANCA disease.
Figures
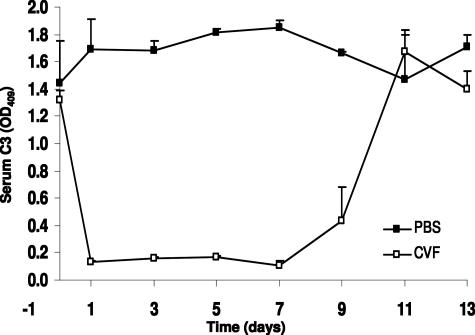
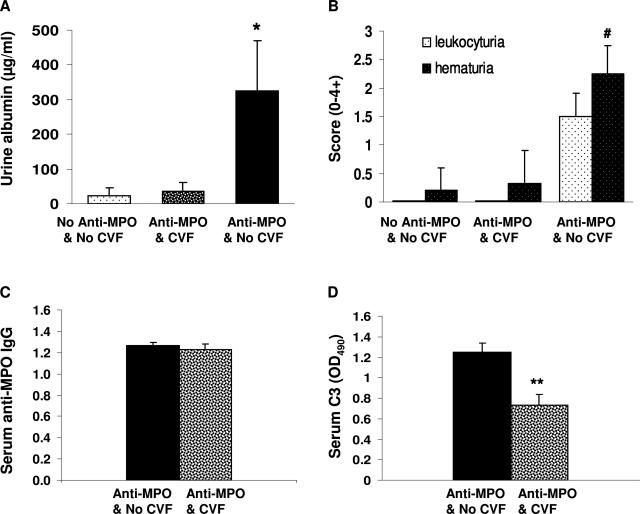
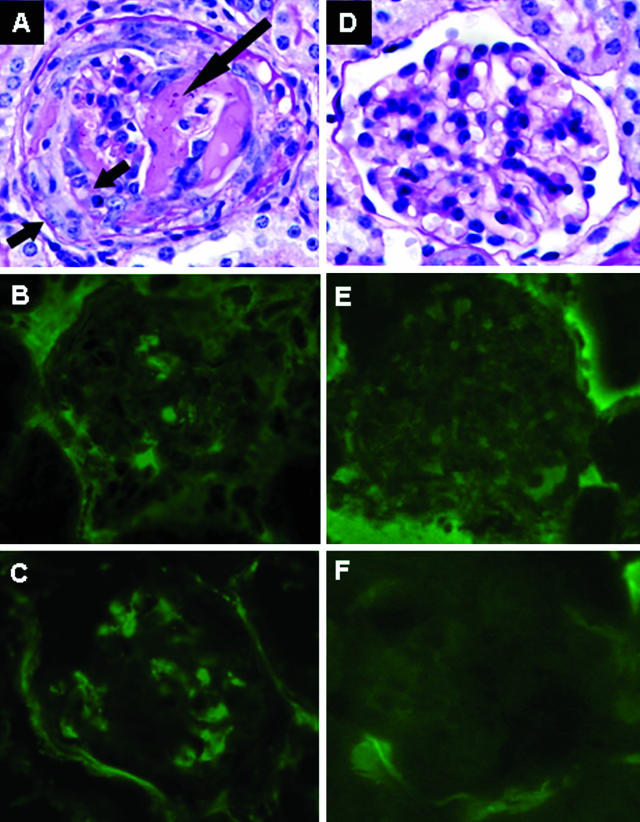
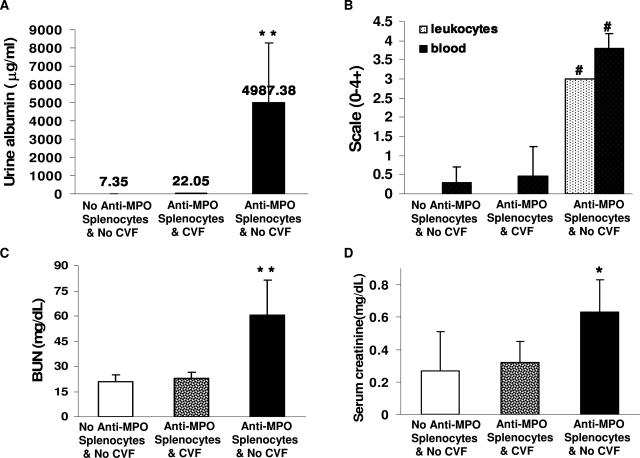
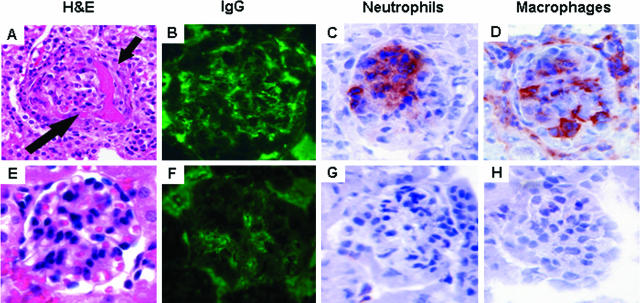
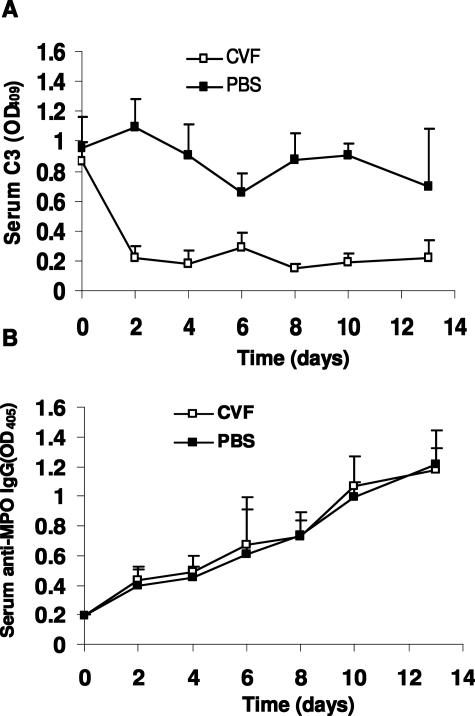
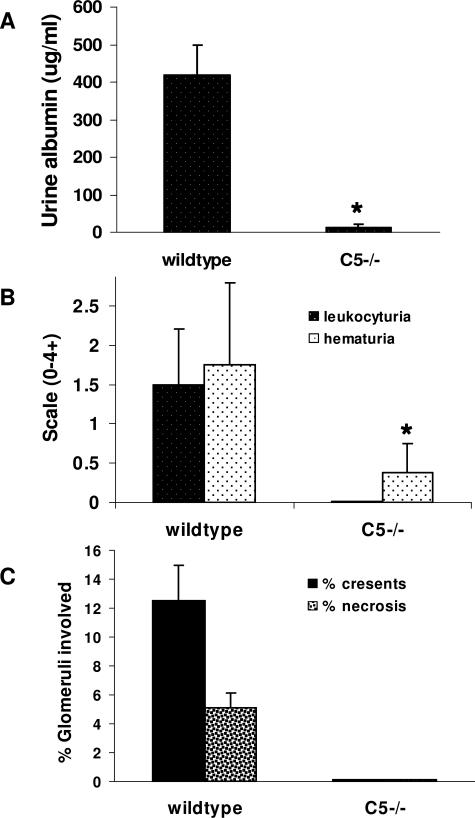
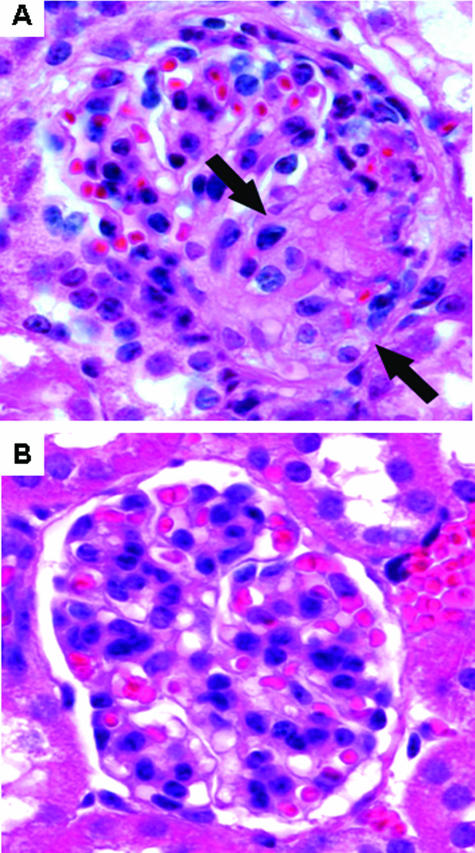
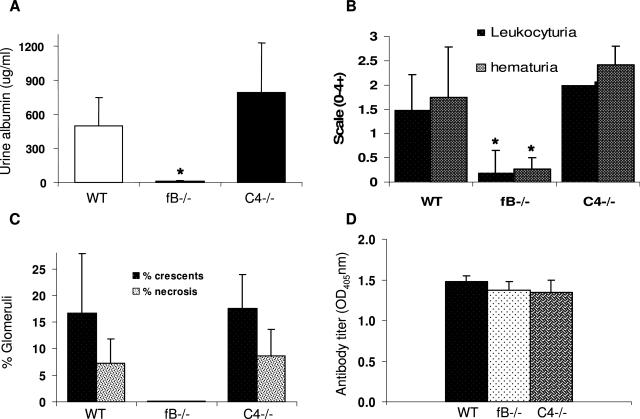
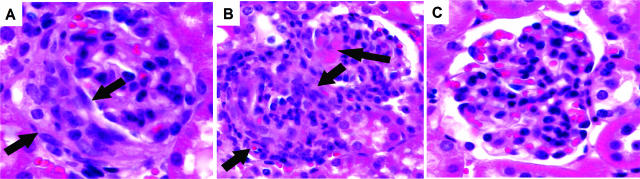
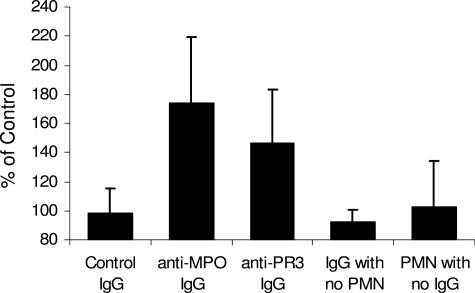
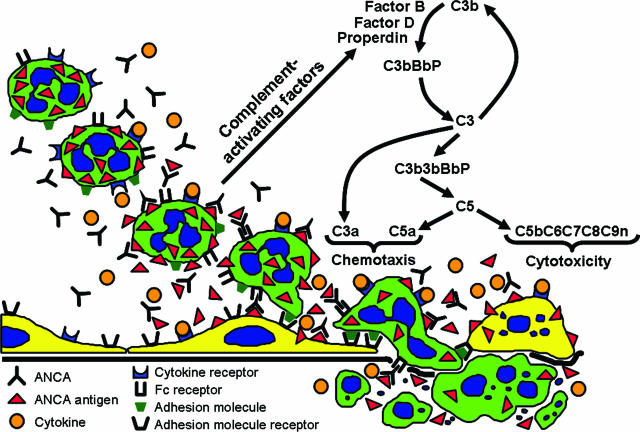
Comment in
-
Alternative complement pathway induction by ANCA.Am J Pathol. 2008 Sep;173(3):910. doi: 10.2353/ajpath.2008.080406. Epub 2008 Aug 7. Am J Pathol. 2008. PMID: 18688036 Free PMC article.
References
-
- Jennette JC, Xiao H, Falk RJ. The pathogenesis of vascular inflammation by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17:1235–1242. - PubMed
-
- Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunology. 2004;93:398–401. - PubMed
-
- Schlieben DJ, Korbet SM, Kimura RE, Schwartz MM, Lewis EJ. Pulmonary-renal syndrome in a newborn with placental transmission of ANCAs. Am J Kidney Dis. 2005;45:758–761. - PubMed
-
- Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, Pusey CD. Anti-neutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005;106:2050–2058. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
