

Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia
- PMID: 17200716
- PMCID: PMC1716209
- DOI: 10.1172/JCI29415
Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia
Abstract
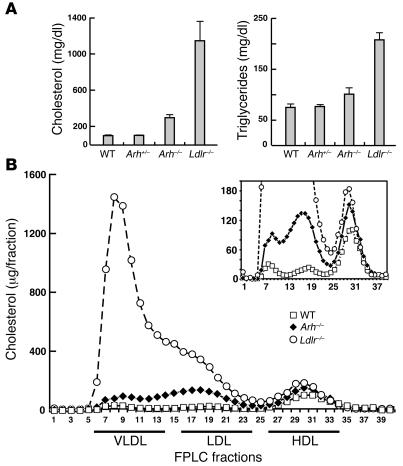
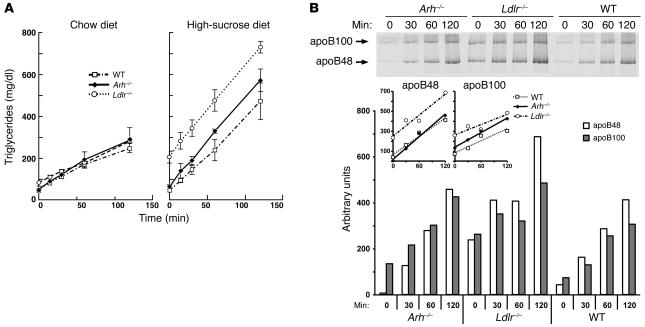
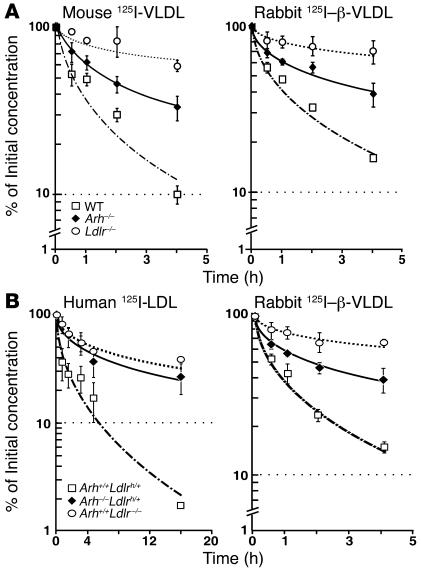
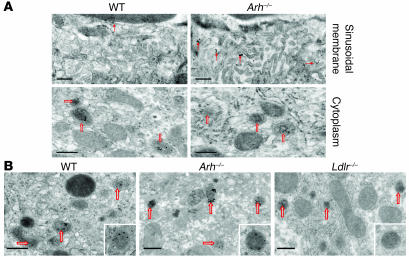
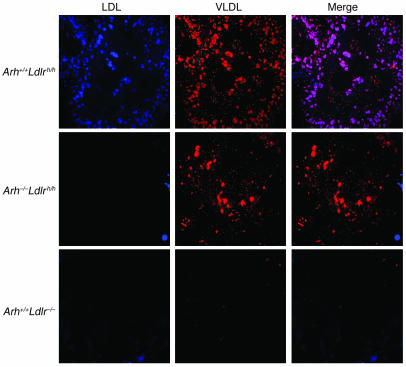
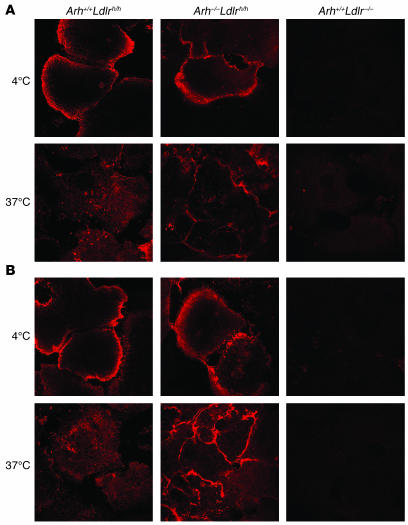
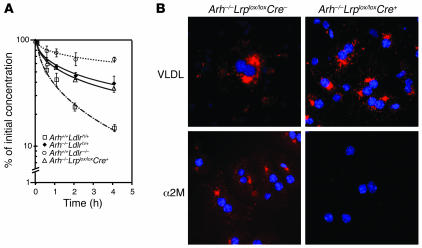
Genetic defects in LDL clearance result in severe hypercholesterolemia and premature atherosclerosis. Mutations in the LDL receptor (LDLR) cause familial hypercholesterolemia (FH), the most severe form of genetic hypercholesterolemia. A phenocopy of FH, autosomal recessive hypercholesterolemia (ARH), is due to mutations in an adaptor protein involved in LDLR internalization. Despite comparable reductions in LDL clearance rates, plasma LDL levels are substantially lower in ARH than in FH. To determine the metabolic basis for this difference, we examined the synthesis and catabolism of VLDL in murine models of FH (Ldlr(-/-)) and ARH (Arh(-/-)). The hyperlipidemic response to a high-sucrose diet was greatly attenuated in Arh(-/-) mice compared with Ldlr(-/-) mice despite similar rates of VLDL secretion. The rate of VLDL clearance was significantly higher in Arh(-/-) mice than in Ldlr(-/-) mice, suggesting that LDLR-dependent uptake of VLDL is maintained in the absence of ARH. Consistent with these findings, hepatocytes from Arh(-/-) mice (but not Ldlr(-/-) mice) internalized beta-migrating VLDL (beta-VLDL). These results demonstrate that ARH is not required for LDLR-dependent uptake of VLDL by the liver. The preservation of VLDL remnant clearance attenuates the phenotype of ARH and likely contributes to greater responsiveness to statins in ARH compared with FH.
Figures
Comment in
-
Atherogenic remnant lipoproteins: role for proteoglycans in trapping, transferring, and internalizing.J Clin Invest. 2007 Jan;117(1):94-8. doi: 10.1172/JCI30889. J Clin Invest. 2007. PMID: 17200713 Free PMC article.
References
-
- Goldstein, J., Hobbs, H., and Brown, M. 2001. Familial hypercholesterolemia. In The metabolic and molecular bases of inherited disease. C. Scriver, A. Beaudet, W. Sly, and D. Valle, editors. McGraw-Hill. New York, New York, USA. 2863–2913.
-
- Khachadurian A.K., Uthman S.M. Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients. Nutr. Metab. 1973;15:132–140. - PubMed
-
- Zuliani G., et al. Severe hypercholesterolaemia: unusual inheritance in an Italian pedigree. Eur. J. Clin. Invest. 1995;25:322–331. - PubMed
-
- Harada-Shiba M., et al. Siblings with normal LDL receptor activity and severe hypercholesterolemia. Arterioscler. Thromb. 1992;12:1071–1078. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
