

Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance
- PMID: 17200721
- PMCID: PMC1716196
- DOI: 10.1172/JCI17645
Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance
Abstract
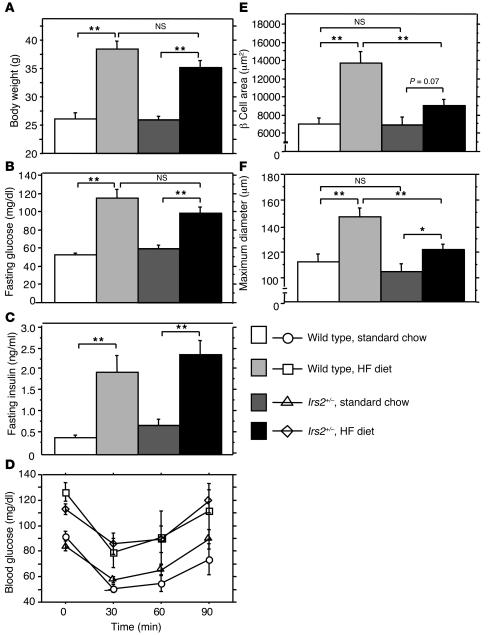
Glucokinase (Gck) functions as a glucose sensor for insulin secretion, and in mice fed standard chow, haploinsufficiency of beta cell-specific Gck (Gck(+/-)) causes impaired insulin secretion to glucose, although the animals have a normal beta cell mass. When fed a high-fat (HF) diet, wild-type mice showed marked beta cell hyperplasia, whereas Gck(+/-) mice demonstrated decreased beta cell replication and insufficient beta cell hyperplasia despite showing a similar degree of insulin resistance. DNA chip analysis revealed decreased insulin receptor substrate 2 (Irs2) expression in HF diet-fed Gck(+/-) mouse islets compared with wild-type islets. Western blot analyses confirmed upregulated Irs2 expression in the islets of HF diet-fed wild-type mice compared with those fed standard chow and reduced expression in HF diet-fed Gck(+/-) mice compared with those of HF diet-fed wild-type mice. HF diet-fed Irs2(+/-) mice failed to show a sufficient increase in beta cell mass, and overexpression of Irs2 in beta cells of HF diet-fed Gck(+/-) mice partially prevented diabetes by increasing beta cell mass. These results suggest that Gck and Irs2 are critical requirements for beta cell hyperplasia to occur in response to HF diet-induced insulin resistance.
Figures







Comment in
-
A dominant role for glucose in beta cell compensation of insulin resistance.J Clin Invest. 2007 Jan;117(1):81-3. doi: 10.1172/JCI30862. J Clin Invest. 2007. PMID: 17200709 Free PMC article.
References
-
- Matschinsky F.M., Ellerman J.E. Metabolism of glucose in the islets of Langerhans. J. Biol. Chem. 1968;243:2730–2736. - PubMed
-
- Velho G., et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40:217–224. - PubMed
-
- Terauchi Y., et al. Pancreatic β-cell-specific targeted disruption of glucokinase gene. J. Biol. Chem. 1995;270:30253–30256. - PubMed
-
- Matschinsky F.M. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223–241. - PubMed
-
- Taylor S.I., Accili D., Imai Y. Insulin resistance or insulin deficiency: which is the primary cause of NIDDM? Diabetes. 1994;43:735–740. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
