

Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury
- PMID: 17200722
- PMCID: PMC1716207
- DOI: 10.1172/JCI29159
Myd88-dependent positioning of Ptgs2-expressing stromal cells maintains colonic epithelial proliferation during injury
Abstract
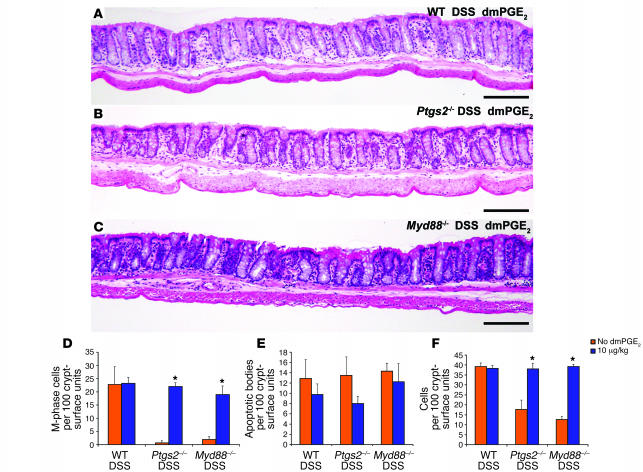
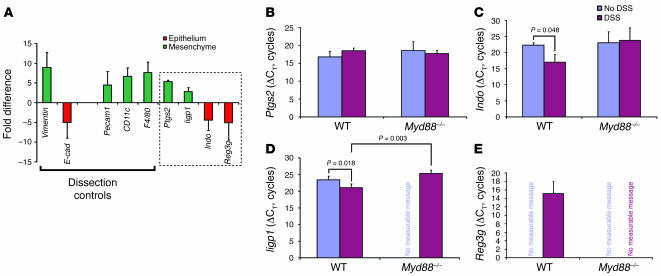
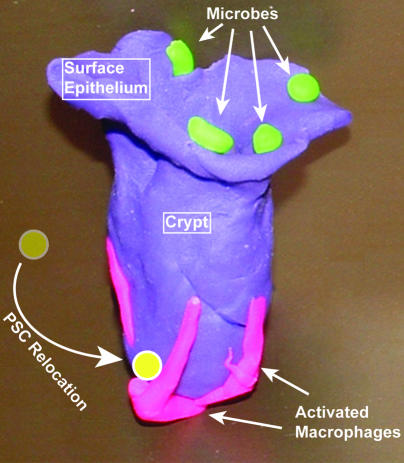
We identified cellular and molecular mechanisms within the stem cell niche that control the activity of colonic epithelial progenitors (ColEPs) during injury. Here, we show that while WT mice maintained ColEP proliferation in the rectum following injury with dextran sodium sulfate, similarly treated Myd88(-/-) (TLR signaling-deficient) and prostaglandin-endoperoxide synthase 2(-/-) (Ptgs2(-/-)) mice exhibited a profound inhibition of epithelial proliferation and cellular organization within rectal crypts. Exogenous addition of 16,16-dimethyl PGE(2) (dmPGE(2)) rescued the effects of this injury in both knockout mouse strains, indicating that Myd88 signaling is upstream of Ptgs2 and PGE(2). In WT and Myd88(-/-) mice, Ptgs2 was expressed in scattered mesenchymal cells. Surprisingly, Ptgs2 expression was not regulated by injury. Rather, in WT mice, the combination of injury and Myd88 signaling led to the repositioning of a subset of the Ptgs2-expressing stromal cells from the mesenchyme surrounding the middle and upper crypts to an area surrounding the crypt base adjacent to ColEPs. These findings demonstrate that Myd88 and prostaglandin signaling pathways interact to preserve epithelial proliferation during injury using what we believe to be a previously undescribed mechanism requiring proper cellular mobilization within the crypt niche.
Figures






Comment in
-
Prostaglandin-secreting cells: a portable first aid kit for tissue repair.J Clin Invest. 2007 Jan;117(1):83-6. doi: 10.1172/JCI30865. J Clin Invest. 2007. PMID: 17200710 Free PMC article.
References
-
- Savage D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977;31:107–133. - PubMed
-
- Dotan, I., and Mayer, L. 2003. Intestinal immunity. In Microbial pathogenesis and the intestinal epithelial cell. G.A. Hecht, editor. ASM Press. Washington, DC, USA. 43–60.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
