

Pancreatic islet cell therapy for type I diabetes: understanding the effects of glucose stimulation on islets in order to produce better islets for transplantation
- PMID: 17201925
- PMCID: PMC1769476
- DOI: 10.1186/1479-5876-5-1
Pancreatic islet cell therapy for type I diabetes: understanding the effects of glucose stimulation on islets in order to produce better islets for transplantation
Abstract
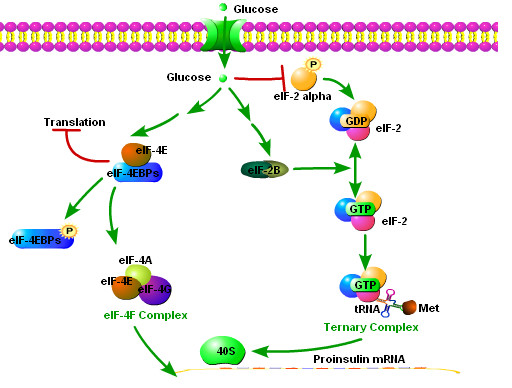
While insulin replacement remains the cornerstone treatment for type I diabetes mellitus (T1DM), the transplantation of pancreatic islets of Langerhans has the potential to become an important alternative. And yet, islet transplant therapy is limited by several factors, including far too few donor pancreases. Attempts to expand mature islets or to produce islets from stem cells are far from clinical application. The production and expansion of the insulin-producing cells within the islet (so called beta cells), or even creating cells that secrete insulin under appropriate physiological control, has proven difficult. The difficulty is explained, in part, because insulin synthesis and release is complex, unique, and not entirely characterized. Understanding beta-cell function at the molecular level will likely facilitate the development of techniques to manufacture beta-cells from stem cells. We will review islet transplantation, as well as the mechanisms underlying insulin transcription, translation and glucose stimulated insulin release.
Figures
Similar articles
-
Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in Type 1 diabetes.Diabetologia. 2004 Apr;47(4):686-94. doi: 10.1007/s00125-004-1372-0. Diabetologia. 2004. PMID: 15298346
-
Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets.Cell Transplant. 2009;18(8):833-45. doi: 10.3727/096368909X471198. Epub 2009 Apr 10. Cell Transplant. 2009. PMID: 19500470
-
Microchip-based engineering of super-pancreatic islets supported by adipose-derived stem cells.Biomaterials. 2014 Jun;35(17):4815-26. doi: 10.1016/j.biomaterials.2014.02.045. Epub 2014 Mar 15. Biomaterials. 2014. PMID: 24636217
-
Human Induced Pluripotent Stem Cells in the Curative Treatment of Diabetes and Potential Impediments Ahead.Adv Exp Med Biol. 2019;1144:25-35. doi: 10.1007/5584_2018_305. Adv Exp Med Biol. 2019. PMID: 30569414 Review.
-
Modulation of the pancreatic islet-stress axis as a novel potential therapeutic target in diabetes mellitus.Vitam Horm. 2014;95:195-222. doi: 10.1016/B978-0-12-800174-5.00008-9. Vitam Horm. 2014. PMID: 24559919 Review.
Cited by
-
Endoplasmic reticulum stress response in an INS-1 pancreatic beta-cell line with inducible expression of a folding-deficient proinsulin.BMC Cell Biol. 2010 Jul 26;11:59. doi: 10.1186/1471-2121-11-59. BMC Cell Biol. 2010. PMID: 20659334 Free PMC article.
-
Improvement of the therapeutic capacity of insulin-producing cells trans-differentiated from human liver cells using engineered cell sheet.Stem Cell Res Ther. 2021 Jan 6;12(1):3. doi: 10.1186/s13287-020-02080-0. Stem Cell Res Ther. 2021. PMID: 33407888 Free PMC article.
-
Efficient and simple production of insulin-producing cells from embryonal carcinoma stem cells using mouse neonate pancreas extract, as a natural inducer.PLoS One. 2014 Mar 10;9(3):e90885. doi: 10.1371/journal.pone.0090885. eCollection 2014. PLoS One. 2014. PMID: 24614166 Free PMC article.
-
Applications of synthetic biology in medical and pharmaceutical fields.Signal Transduct Target Ther. 2023 May 11;8(1):199. doi: 10.1038/s41392-023-01440-5. Signal Transduct Target Ther. 2023. PMID: 37169742 Free PMC article. Review.
-
Stem Cell-Derived Exosomes as a Therapeutic Option for Spinal Cord Injuries; a Systematic Review and Meta-Analysis.Arch Acad Emerg Med. 2024 Sep 5;13(1):e2. doi: 10.22037/aaem.v12i1.2261. eCollection 2025. Arch Acad Emerg Med. 2024. PMID: 39318865 Free PMC article. Review.
References
-
- Steffes MW, Sibley S, Jackson M, Thomas W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care. 2003;26:832–836. - PubMed
-
- Gruessner AC, Sutherland DE. Pancreas transplant outcomes for United States (US) and non-US cases as reported to the United Network for Organ Sharing (UNOS) and the International Pancreas Transplant Registry (IPTR) as of June 2004. Clin Transplant. 2005;19:433–455. doi: 10.1111/j.1399-0012.2005.00378.x. - DOI - PubMed
-
- Coppelli A, Giannarelli R, Vistoli F, Del Prato S, Rizzo G, Mosca F, Boggi U, Marchetti P. The beneficial effects of pancreas transplant alone on diabetic nephropathy. Diabetes Care. 2005;28:1366–1370. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
