

Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization
- PMID: 17210698
- PMCID: PMC2941512
- DOI: 10.1158/0008-5472.CAN-06-3292
Amplification of tumor hypoxic responses by macrophage migration inhibitory factor-dependent hypoxia-inducible factor stabilization
Abstract
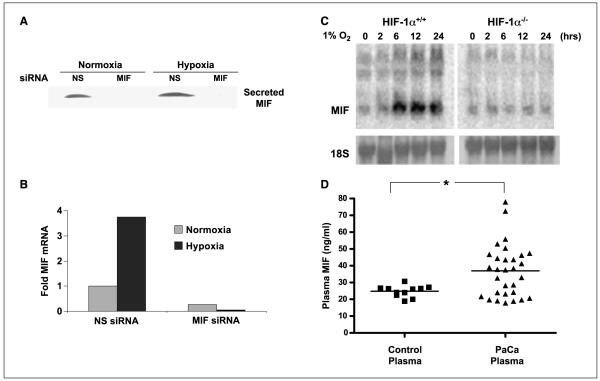
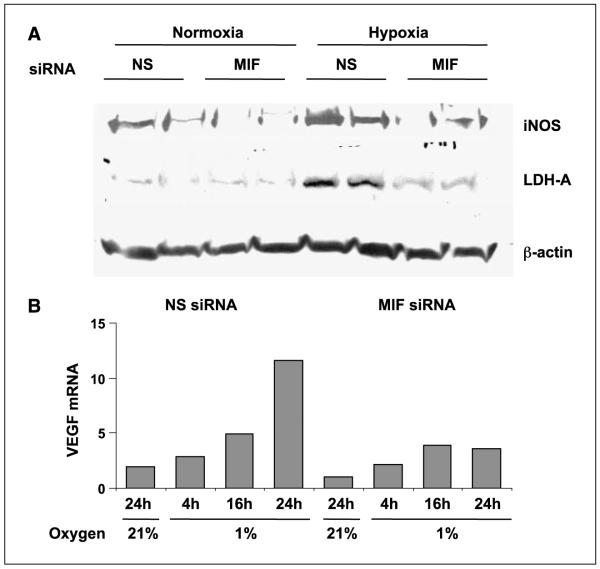
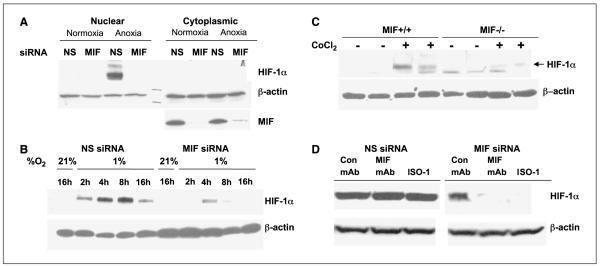
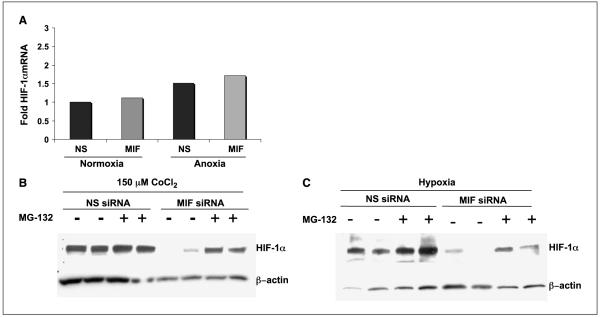
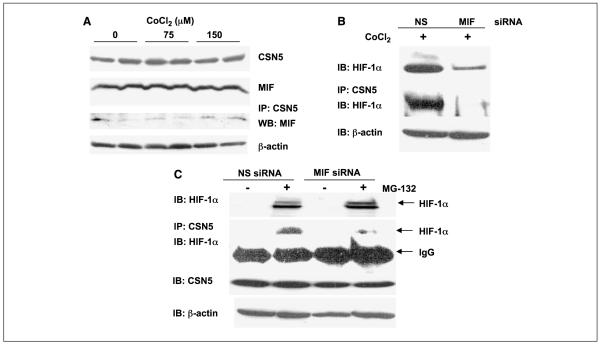
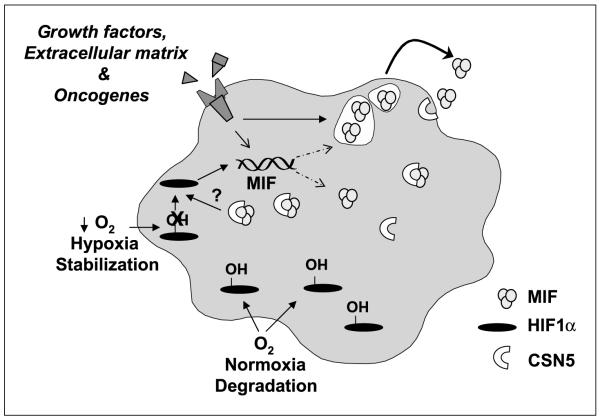
Low oxygen tension-mediated transcription by hypoxia-inducible factors (HIF) has been reported to facilitate tumor progression, therapeutic resistance, and metastatic adaptation. One previously described target of hypoxia-mediated transcription is the cytokine/growth factor macrophage migration inhibitory factor (MIF). In studies designed to better understand hypoxia-stimulated MIF function, we have discovered that not only is MIF induced by hypoxia in pancreatic adenocarcinoma but MIF is also necessary for maximal hypoxia-induced HIF-1alpha expression. Cells lacking MIF are defective in hypoxia- and prolyl hydroxylase inhibitor-induced HIF-1alpha stabilization and subsequent transcription of glycolytic and angiogenic gene products. Moreover, COP9 signalosome subunit 5 (CSN5), a component of the COP9 signalosome previously reported to functionally interact with MIF, has recently been shown to interact with and stabilize HIF-1alpha. Our results indicate that MIF interacts with CSN5 in pancreatic cancer cells and that MIF-depleted cells display marked defects in hypoxia-induced CSN5/HIF-1alpha interactions. This functional interdependence between HIF-1alpha and MIF may represent an important and previously unrecognized pro-tumorigenic axis.
Figures
Similar articles
-
Macrophage migration inhibitory factor manipulation and evaluation in tumoral hypoxic adaptation.Methods Enzymol. 2007;435:355-69. doi: 10.1016/S0076-6879(07)35018-0. Methods Enzymol. 2007. PMID: 17998063 Free PMC article. Review.
-
HIF1α-Induced by Lysophosphatidic Acid Is Stabilized via Interaction with MIF and CSN5.PLoS One. 2015 Sep 9;10(9):e0137513. doi: 10.1371/journal.pone.0137513. eCollection 2015. PLoS One. 2015. PMID: 26352431 Free PMC article.
-
Unraveling the role of hypoxia-inducible factor (HIF)-1α and HIF-2α in the adaption process of human microvascular endothelial cells (HMEC-1) to hypoxia: Redundant HIF-dependent regulation of macrophage migration inhibitory factor.Microvasc Res. 2018 Mar;116:34-44. doi: 10.1016/j.mvr.2017.09.004. Epub 2017 Oct 6. Microvasc Res. 2018. PMID: 28993199
-
Macrophage migration inhibitory factor is regulated by HIF-1α and cAMP and promotes renal cyst cell proliferation in a macrophage-independent manner.J Mol Med (Berl). 2020 Nov;98(11):1547-1559. doi: 10.1007/s00109-020-01964-1. Epub 2020 Sep 4. J Mol Med (Berl). 2020. PMID: 32885302 Free PMC article.
-
COPing with hypoxia.Semin Cell Dev Biol. 2005 Aug-Oct;16(4-5):462-73. doi: 10.1016/j.semcdb.2005.03.002. Semin Cell Dev Biol. 2005. PMID: 15916908 Free PMC article. Review.
Cited by
-
Macrophage migration inhibitory factor produced by the tumour stroma but not by tumour cells regulates angiogenesis in the B16-F10 melanoma model.Br J Cancer. 2012 Oct 23;107(9):1498-505. doi: 10.1038/bjc.2012.392. Epub 2012 Sep 6. Br J Cancer. 2012. PMID: 22955855 Free PMC article.
-
Immunopathophysiology of Juvenile Spondyloarthritis (jSpA): The "Out of the Box" View on Epigenetics, Neuroendocrine Pathways and Role of the Macrophage Migration Inhibitory Factor (MIF).Front Med (Lausanne). 2021 Oct 6;8:700982. doi: 10.3389/fmed.2021.700982. eCollection 2021. Front Med (Lausanne). 2021. PMID: 34692718 Free PMC article. Review.
-
Overexpression of macrophage migration inhibitory factor in adenoid cystic carcinoma: correlation with enhanced metastatic potential.J Cancer Res Clin Oncol. 2013 Feb;139(2):287-95. doi: 10.1007/s00432-012-1330-z. Epub 2012 Oct 12. J Cancer Res Clin Oncol. 2013. PMID: 23064787 Free PMC article.
-
Targeting macrophage migration inhibitory factor as a potential therapeutic strategy in colorectal cancer.Oncogenesis. 2025 Aug 20;14(1):30. doi: 10.1038/s41389-025-00572-3. Oncogenesis. 2025. PMID: 40835826 Free PMC article.
-
Macrophage migration inhibitory factor (MIF) in the development and progression of pulmonary arterial hypertension.Glob Cardiol Sci Pract. 2018 Jun 30;2018(2):14. doi: 10.21542/gcsp.2018.14. Glob Cardiol Sci Pract. 2018. PMID: 30083544 Free PMC article. Review.
References
-
- Maxwell PH. The HIF pathway in cancer. Semin Cell Dev Biol. 2005;16:523–30. - PubMed
-
- Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol. 2002;13:29–37. - PubMed
-
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. - PubMed
-
- Brown JM. Tumor microenvironment and the response to anticancer therapy. Cancer Biol Ther. 2002;1:453–8. - PubMed
-
- Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5:429–41. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
