

Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma
- PMID: 17210792
- PMCID: PMC1759904
- DOI: 10.1101/gad.1480007
Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma
Erratum in
- Genes Dev. 2007 Feb 15;21(4):481
Abstract
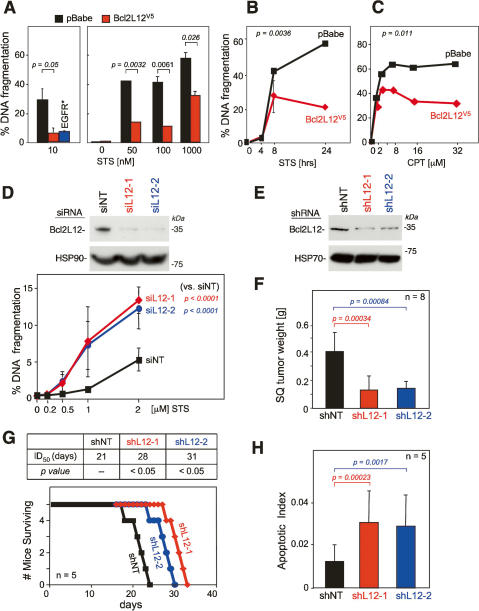
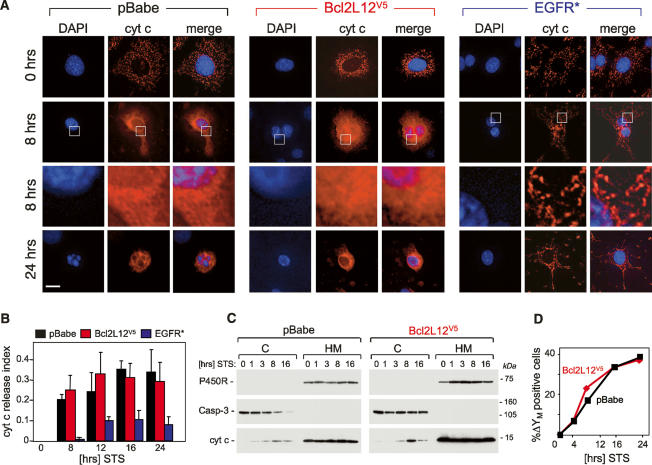
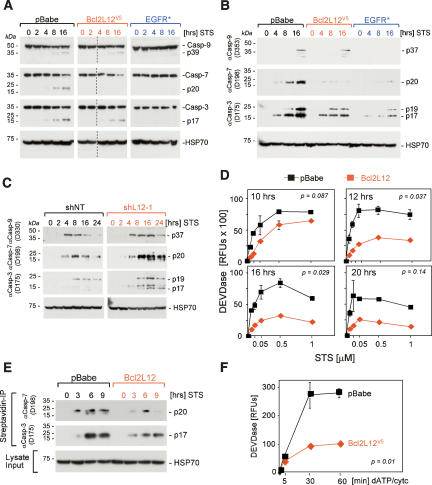
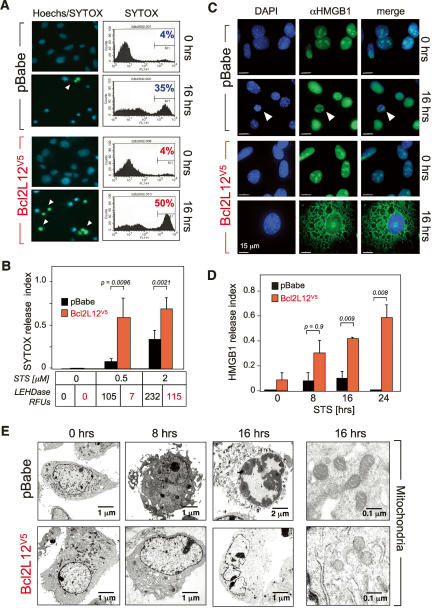
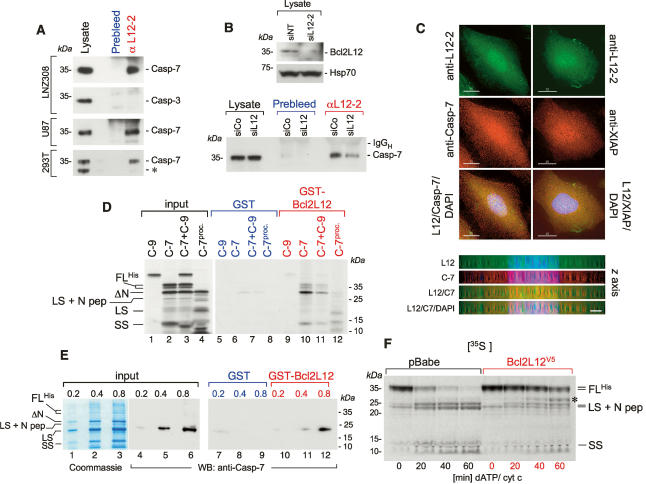
Glioblastoma (GBM) is an astrocytic brain tumor characterized by an aggressive clinical course and intense resistance to all therapeutic modalities. Here, we report the identification and functional characterization of Bcl2L12 (Bcl2-like-12) that is robustly expressed in nearly all human primary GBMs examined. Enforced Bcl2L12 expression confers marked apoptosis resistance in primary cortical astrocytes, and, conversely, its RNA interference (RNAi)-mediated knockdown sensitizes human glioma cell lines toward apoptosis in vitro and impairs tumor growth with increased intratumoral apoptosis in vivo. Mechanistically, Bcl2L12 expression does not affect cytochrome c release or apoptosome-driven caspase-9 activation, but instead inhibits post-mitochondrial apoptosis signaling at the level of effector caspase activation. One of Bcl2L12's mechanisms of action stems from its ability to interact with and neutralize caspase-7. Notably, while enforced Bcl2L12 expression inhibits apoptosis, it also engenders a pronecrotic state, which mirrors the cellular phenotype elicited by genetic or pharmacologic inhibition of post-mitochondrial apoptosis molecules. Thus, Bcl2L12 contributes to the classical tumor biological features of GBM such as intense apoptosis resistance and florid necrosis, and may provide a target for enhanced therapeutic responsiveness of this lethal cancer.
Figures
References
-
- Aoyama A., Steiger R.H., Frohli E., Schafer R., von Deimling A., Wiestler O.D., Klemenz R., Steiger R.H., Frohli E., Schafer R., von Deimling A., Wiestler O.D., Klemenz R., Frohli E., Schafer R., von Deimling A., Wiestler O.D., Klemenz R., Schafer R., von Deimling A., Wiestler O.D., Klemenz R., von Deimling A., Wiestler O.D., Klemenz R., Wiestler O.D., Klemenz R., Klemenz R. 1993. Expression of α B-crystallin in human brain tumors. Int. J. Cancer. 1993;55:760–764. - PubMed
-
- Brat D.J., Van Meir E.G., Van Meir E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Invest. 2004;84:397–405. - PubMed
-
- Denault J.B., Bekes M., Scott F.L., Sexton K.M., Bogyo M., Salvesen G.S., Bekes M., Scott F.L., Sexton K.M., Bogyo M., Salvesen G.S., Scott F.L., Sexton K.M., Bogyo M., Salvesen G.S., Sexton K.M., Bogyo M., Salvesen G.S., Bogyo M., Salvesen G.S., Salvesen G.S. Engineered hybrid dimers: Tracking the activation pathway of caspase-7. Mol. Cell. 2006;23:523–533. - PubMed
-
- Ferri K.F., Kroemer G., Kroemer G. Mitochondria—The suicide organelles. Bioessays. 2001;23:111–115. - PubMed
-
- Fujita E., Egashira J., Urase K., Kuida K., Momoi T., Egashira J., Urase K., Kuida K., Momoi T., Urase K., Kuida K., Momoi T., Kuida K., Momoi T., Momoi T. Caspase-9 processing by caspase-3 via a feedback amplification loop in vivo. Cell Death Differ. 2001;8:335–344. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources