

The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease
- PMID: 17220890
- PMCID: PMC2657343
- DOI: 10.1038/ng1943
The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease
Abstract
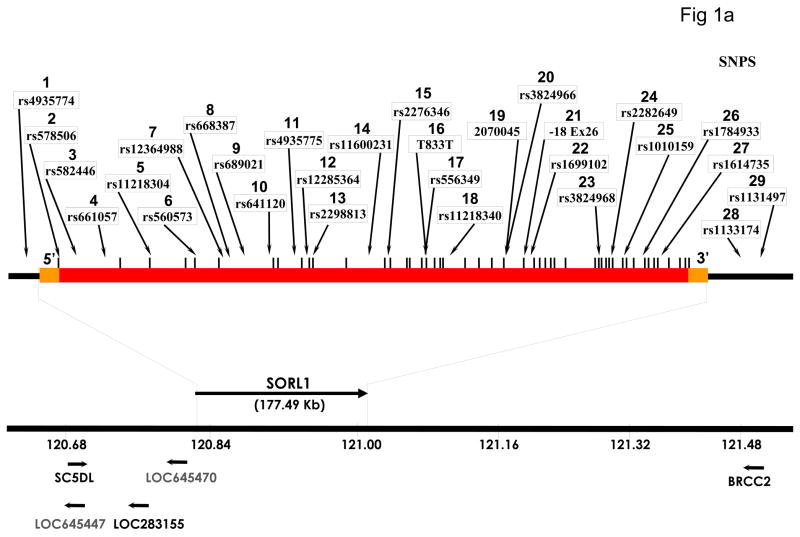
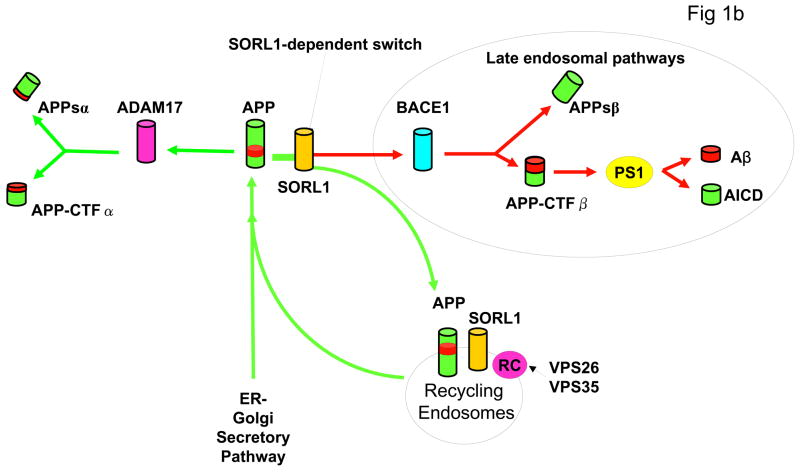
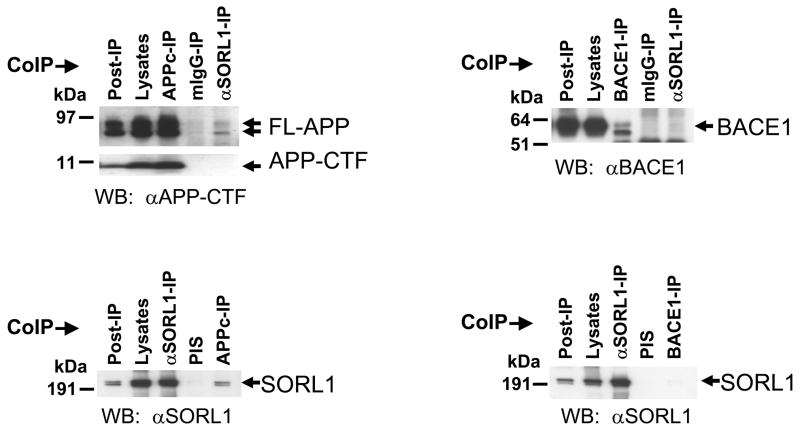
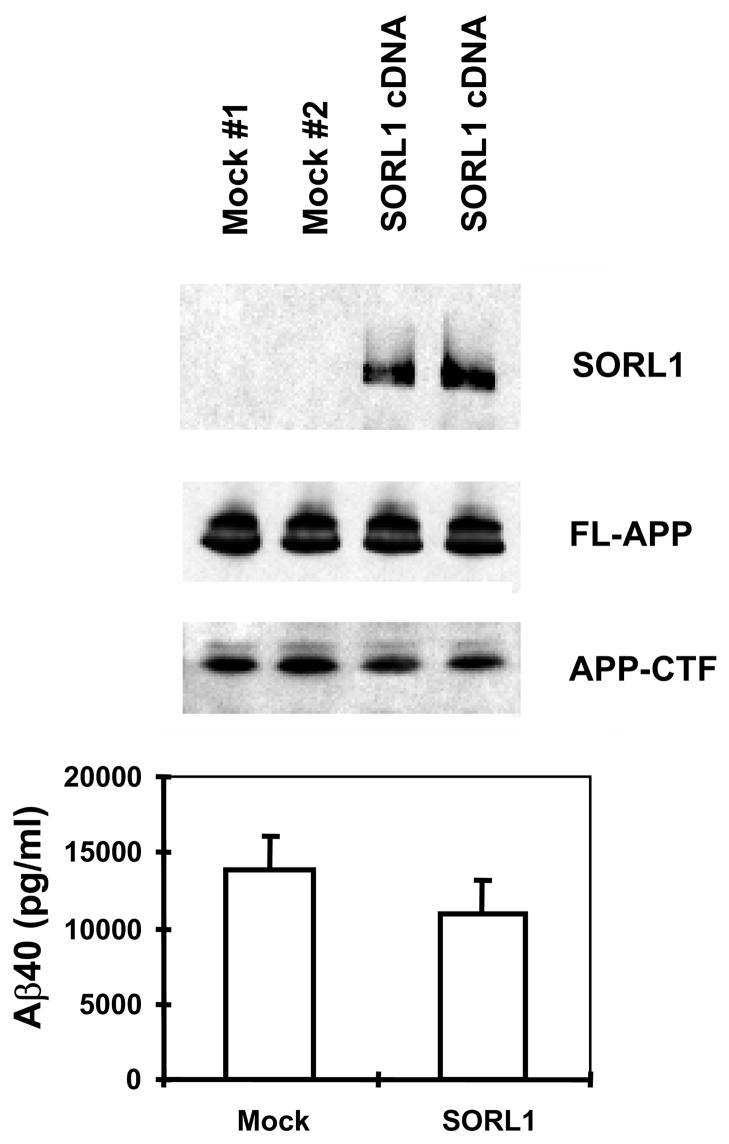
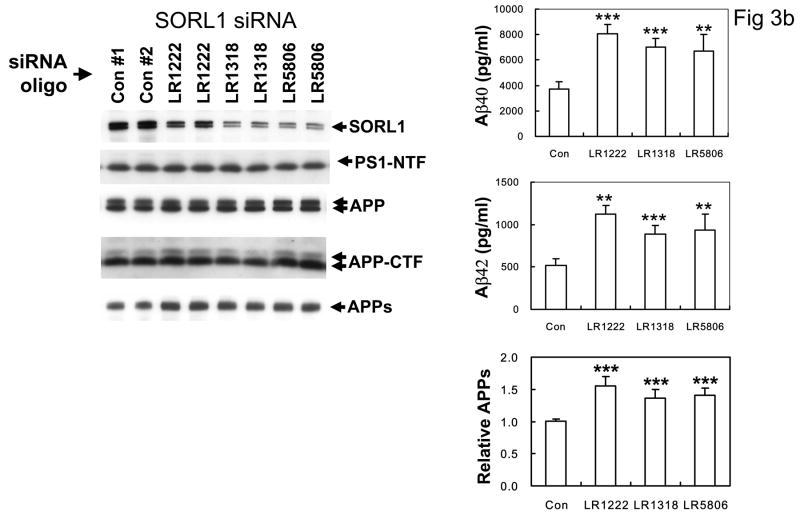
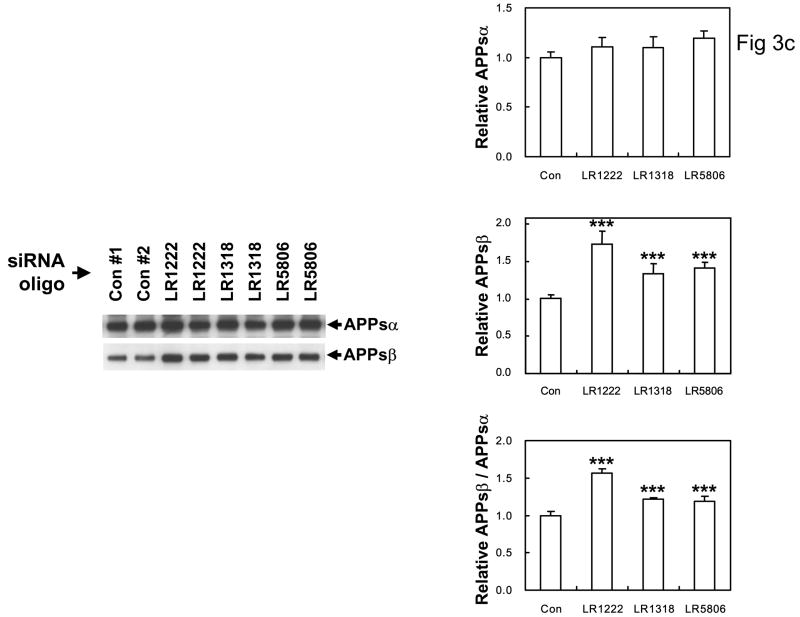
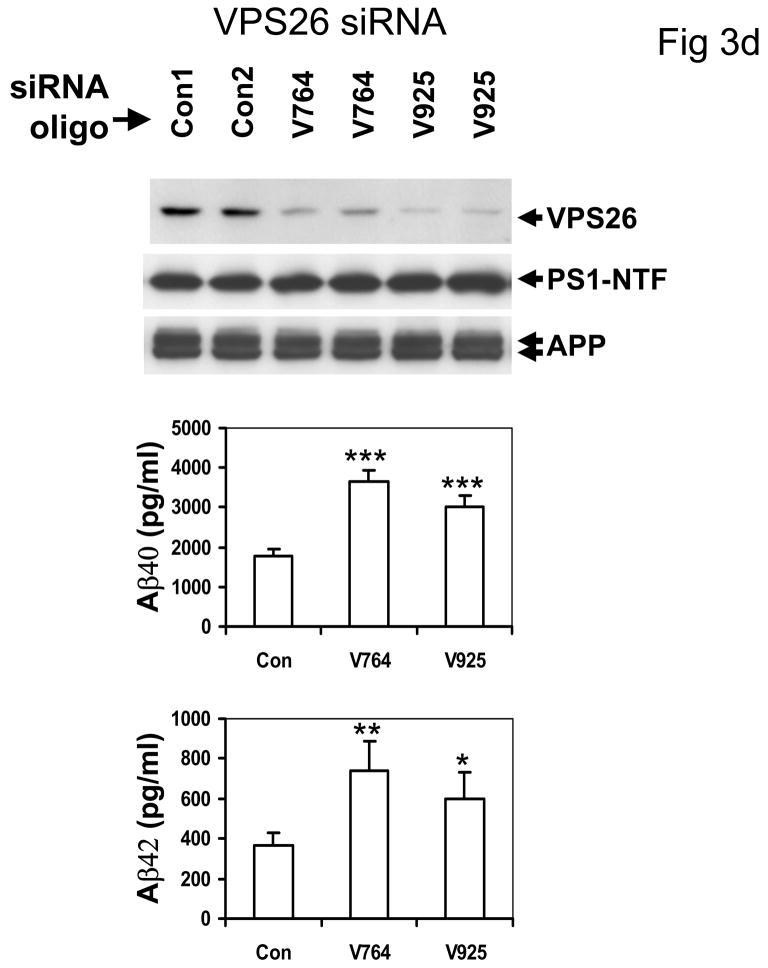
The recycling of the amyloid precursor protein (APP) from the cell surface via the endocytic pathways plays a key role in the generation of amyloid beta peptide (Abeta) in Alzheimer disease. We report here that inherited variants in the SORL1 neuronal sorting receptor are associated with late-onset Alzheimer disease. These variants, which occur in at least two different clusters of intronic sequences within the SORL1 gene (also known as LR11 or SORLA) may regulate tissue-specific expression of SORL1. We also show that SORL1 directs trafficking of APP into recycling pathways and that when SORL1 is underexpressed, APP is sorted into Abeta-generating compartments. These data suggest that inherited or acquired changes in SORL1 expression or function are mechanistically involved in causing Alzheimer disease.
Conflict of interest statement
The authors have no competing financial interests.
Figures
References
-
- Goate AM, et al. Segregation of a missense mutation in the amyloid precursor protein gene with Familial Alzheimer Disease. Nature. 1991;349:704–706. - PubMed
-
- Sherrington R, et al. Cloning of a gene bearing missense mutations in early onset familial Alzheimer’s disease. Nature. 1995;375:754–760. - PubMed
-
- Rogaev EI, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a novel gene on chromosome 1 related to the Alzheimer’s Disease type 3 gene. Nature. 1995;376:775–778. - PubMed
-
- Saunders A, et al. Association of Apoliprotein E allele e4 with the late-onset familial and sporadic Alzheimer Disease. Neurology. 1993;43:1467–1472. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AG009029/AG/NIA NIH HHS/United States
- P50 AG016574/AG/NIA NIH HHS/United States
- P01-AG07232/AG/NIA NIH HHS/United States
- P30 AG013846/AG/NIA NIH HHS/United States
- P01 AG007232/AG/NIA NIH HHS/United States
- P50-AG16574/AG/NIA NIH HHS/United States
- R37 AG015473/AG/NIA NIH HHS/United States
- R37-AG15473/AG/NIA NIH HHS/United States
- R01 AG017173/AG/NIA NIH HHS/United States
- U01-AG06786/AG/NIA NIH HHS/United States
- U01 AG006786/AG/NIA NIH HHS/United States
- R01-HG/AG02213/AG/NIA NIH HHS/United States
- R01-AG09029/AG/NIA NIH HHS/United States
- R01-AG017173/AG/NIA NIH HHS/United States
- P30-AG13846/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
