

Viral determinants of resistance to treatment in patients with hepatitis C
- PMID: 17223621
- PMCID: PMC1797633
- DOI: 10.1128/CMR.00010-06
Viral determinants of resistance to treatment in patients with hepatitis C
Abstract
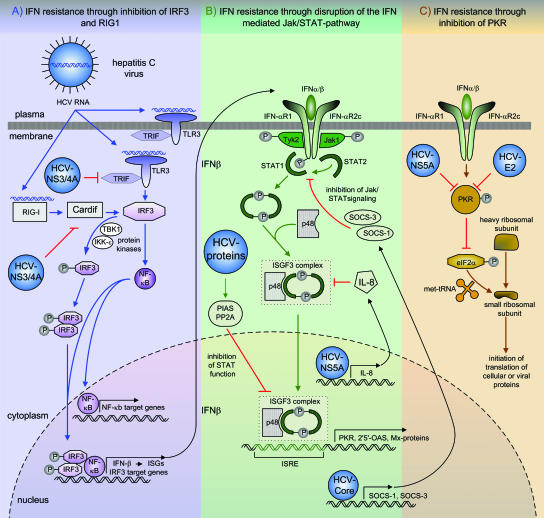
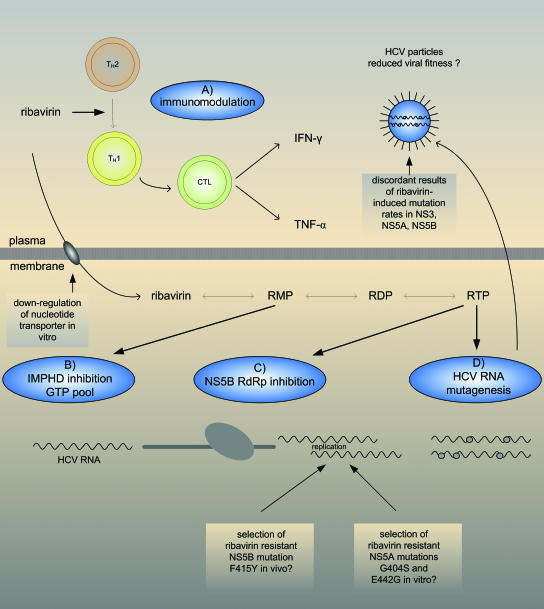
Chronic hepatitis C virus (HCV) infection affects more than 170 million persons worldwide and is responsible for the development of liver cirrhosis in many cases. Standard treatment with pegylated alpha interferon (IFN-alpha) in combination with the nucleoside analogue ribavirin leads to a sustained virologic response in approximately half of the patients. IFN-alpha is classified as an indirect treatment, as it interacts with the host's immune response. The mechanism of action of ribavirin is still unknown. The benefit of triple therapy by adding other antiviral agents, e.g., amantadine, is controversial. Currently, new direct antiviral drugs (HCV protease/polymerase inhibitors) are being evaluated in phase 1/phase 2 trials. Phenotypic resistance to antiviral therapy has been attributed to amino acid variations within distinct regions of the HCV polyprotein. While sensitivity to IFN-alpha-based antiviral therapy in vivo is clearly correlated with the number of mutations within the HCV NS5A protein, the underlying functional mechanisms for this association are unknown. In turn, in vitro, several mechanisms to circumvent the host immune defense or to block treatment-induced antiviral activities have been described for different HCV proteins. By the introduction of direct antiviral drugs, hepatitis C therapy now is entering a new era in which the development of resistance may become the most important parameter for treatment success or failure.
Figures
References
-
- Abbate, I., I. Lo Iacono, R. Di Stefano, G. Cappiello, E. Girardi, R. Longo, D. Ferraro, G. Antonucci, V. Di Marco, M. Solmone, A. Craxi, G. Ippolito, and M. R. Capobianchi. 2004. HVR-1 quasispecies modifications occur early and are correlated to initial but not sustained response in HCV-infected patients treated with pegylated- or standard-interferon and ribavirin. J. Hepatol. 40:831-836. - PubMed
-
- Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732-738. - PubMed
-
- Andant, C., J. Lamoril, J. C. Deybach, P. Jouet, and J. C. Soule. 2000. Amantadine for chronic hepatitis C: pilot study in 14 patients. Eur. J. Gastroenterol. Hepatol. 12:1319-1322. - PubMed
-
- Asahina, Y., N. Izumi, N. Enomoto, M. Uchihara, M. Kurosaki, Y. Onuki, Y. Nishimura, K. Ueda, K. Tsuchiya, H. Nakanishi, T. Kitamura, and S. Miyake. 2005. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 43:623-629. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
