

Epidermal growth factor receptor signaling is required for microadenoma formation in the mouse azoxymethane model of colonic carcinogenesis
- PMID: 17234795
- PMCID: PMC2705749
- DOI: 10.1158/0008-5472.CAN-05-3343
Epidermal growth factor receptor signaling is required for microadenoma formation in the mouse azoxymethane model of colonic carcinogenesis
Abstract
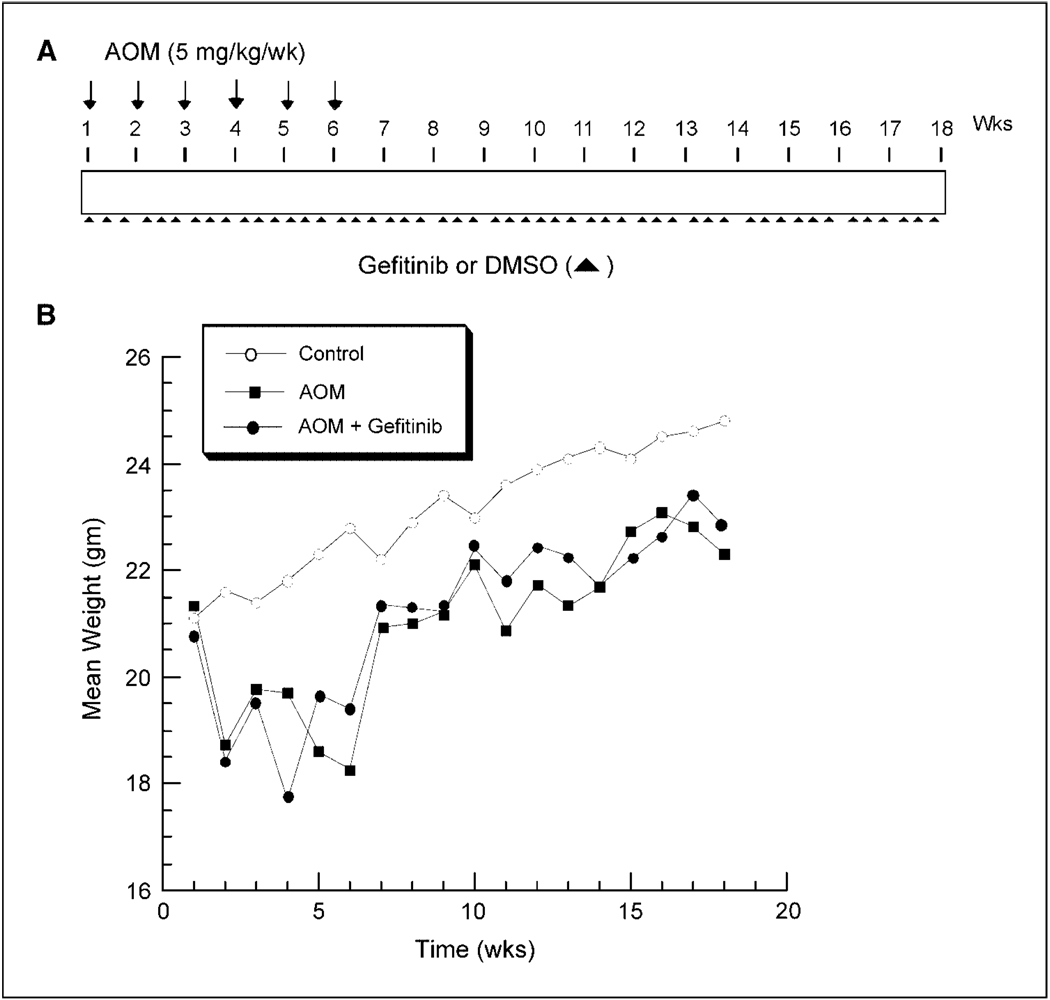
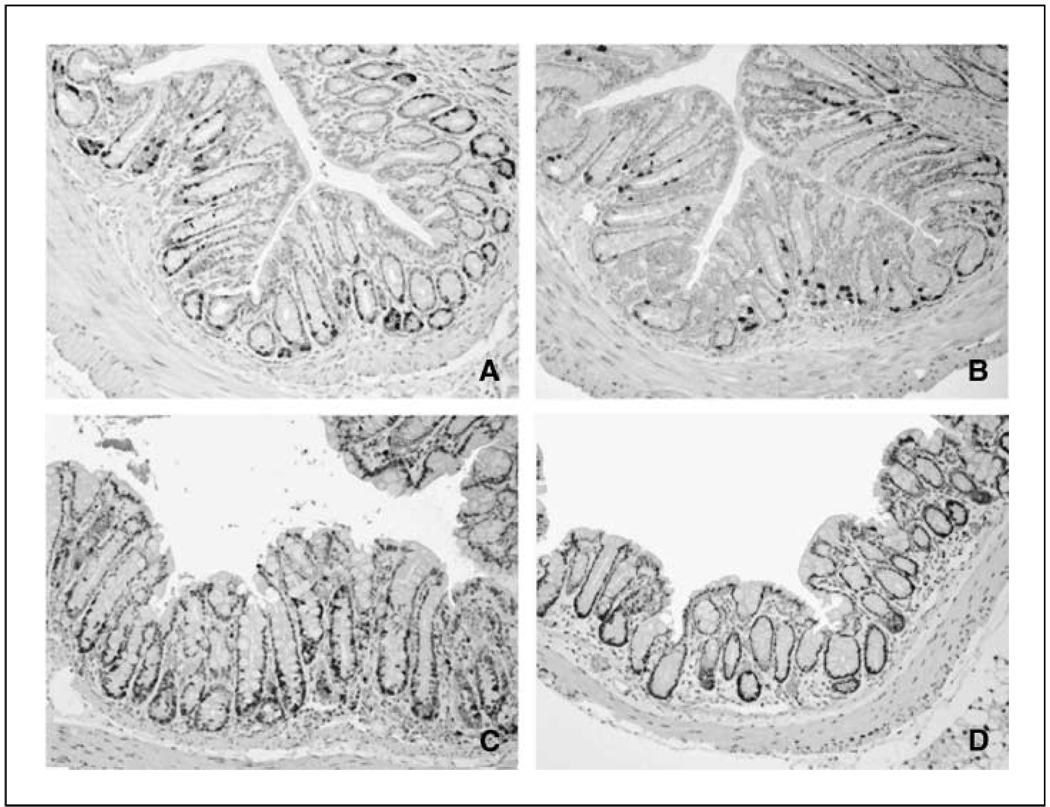
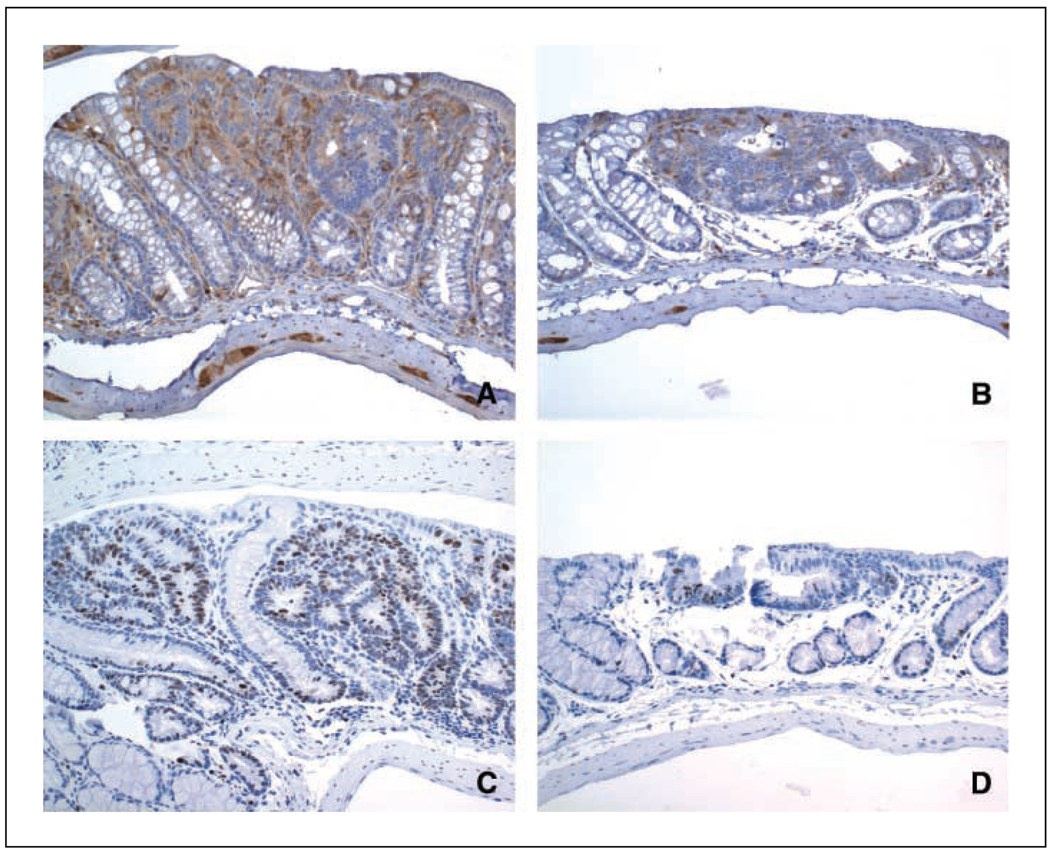
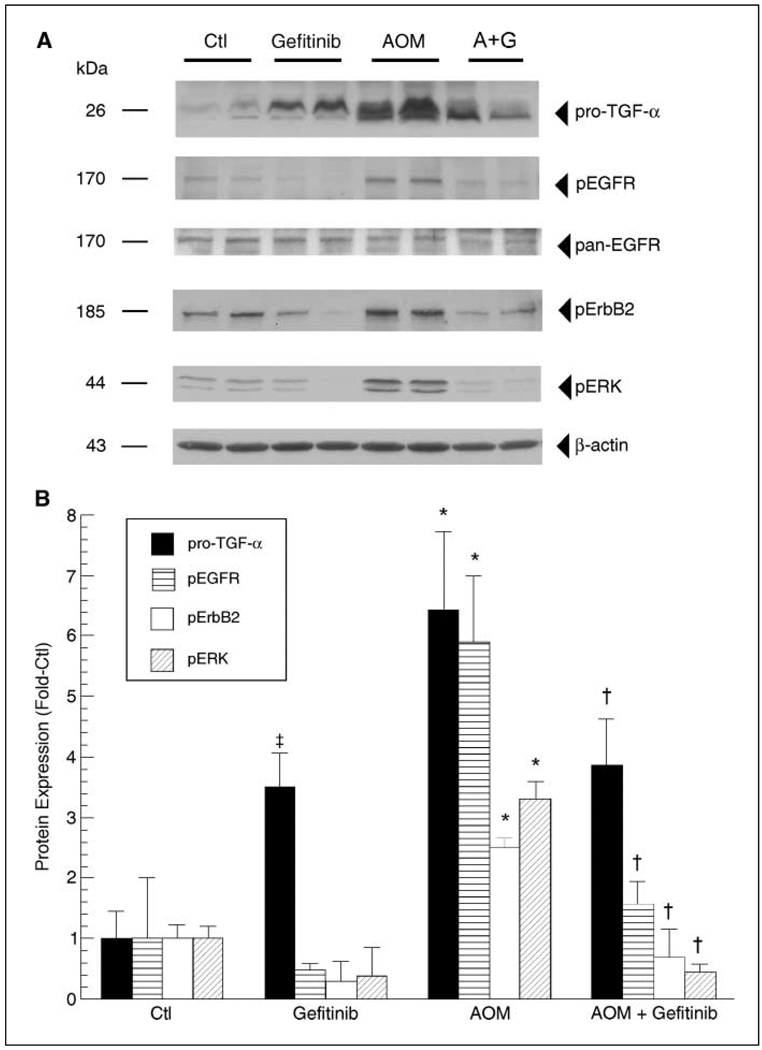
Colonic carcinogenesis involves the progressive dysregulation of homeostatic mechanisms that control growth. The epidermal growth factor (EGF) receptor (EGFR) regulates colonocyte growth and differentiation and is overexpressed in many human colon cancers. A requirement for EGFR in colonic premalignancy, however, has not been shown. In the current study, we used a specific EGFR antagonist, gefitinib, to investigate this role of the receptor in azoxymethane colonic premalignancy. The azoxymethane model shares many clinical, histologic, and molecular features of human colon cancer. Mice received azoxymethane i.p. (5 mg/kg/wk) or saline for 6 weeks. Animals were also gavaged with gefitinib (10 mg/kg body weight) or vehicle (DMSO) thrice weekly for 18 weeks, a dose schedule that inhibited normal receptor activation by exogenous EGF. Compared with control colonocytes [bromodeoxyuridine (BrdUrd), 2.2+/-1.2%], azoxymethane significantly increased proliferation (BrdUrd, 12.6+/-2.8%), whereas gefitinib inhibited this hyperproliferation (BrdUrd, 6.2+/-4.0%; <0.005). Azoxymethane significantly induced pro-transforming growth factor-alpha (6.4+/-1.3-fold) and increased phospho-(active) EGFR (5.9+/-1.1-fold), phospho-(active) ErbB2 (2.3+/-0.2-fold), and phospho-(active) extracellular signal-regulated kinase (3.3+/-0.4-fold) in premalignant colonocytes. Gefitinib inhibited activations of these kinases by >75% (P<0.05). Gefitinib also significantly reduced the number of large aberrant crypt foci and decreased the incidence of colonic microadenomas from 75% to 33% (P<0.05). Gefitinib concomitantly decreased cell cycle-regulating cyclin D1 and prostanoid biosynthetic enzyme cyclooxygenase-2 in microadenomas, suggesting that these regulators are key targets of EGFR in colonic carcinogenesis. These results show for the first time that EGFR signaling is required for early stages of colonic carcinogenesis. Our findings suggest, moreover, that inhibitors of EGFR might be useful in chemopreventive strategies in individuals at increased risk for colonic malignancies.
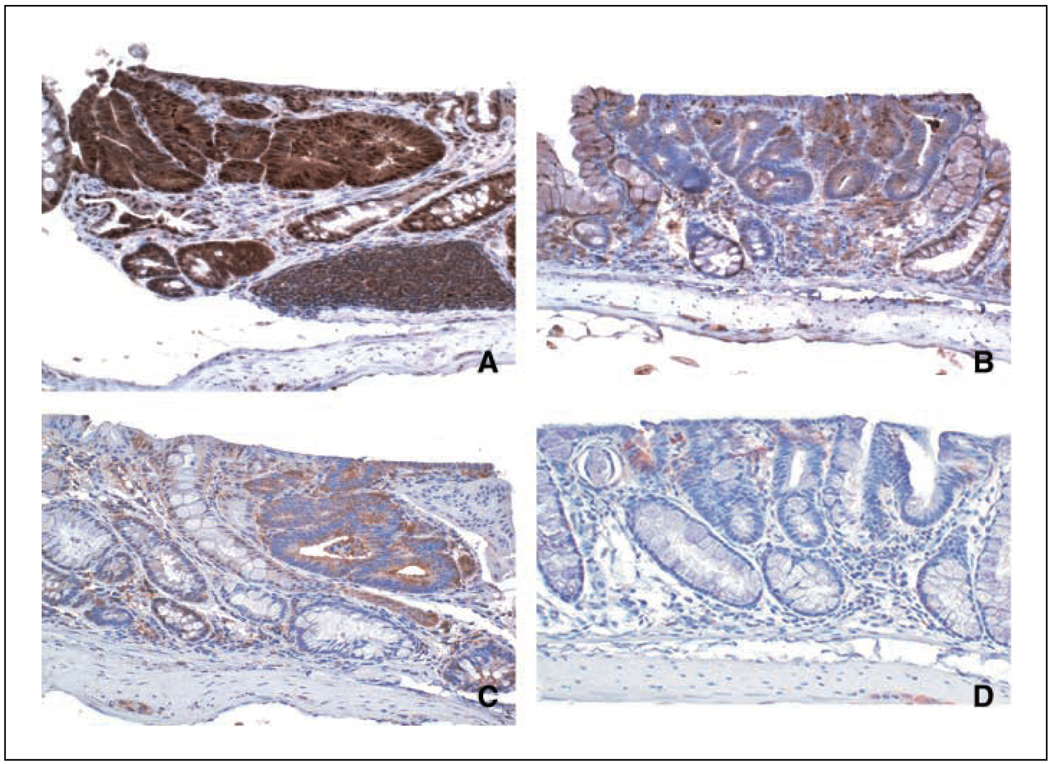
Figures
Similar articles
-
Epidermal growth factor receptor controls flat dysplastic aberrant crypt foci development and colon cancer progression in the rat azoxymethane model.Clin Cancer Res. 2008 Apr 15;14(8):2253-62. doi: 10.1158/1078-0432.CCR-07-4926. Clin Cancer Res. 2008. PMID: 18413814
-
Ursodeoxycholic acid and F(6)-D(3) inhibit aberrant crypt proliferation in the rat azoxymethane model of colon cancer: roles of cyclin D1 and E-cadherin.Cancer Epidemiol Biomarkers Prev. 2002 Dec;11(12):1653-62. Cancer Epidemiol Biomarkers Prev. 2002. PMID: 12496057
-
Altered expression of beta-catenin, inducible nitric oxide synthase and cyclooxygenase-2 in azoxymethane-induced rat colon carcinogenesis.Carcinogenesis. 2000 Jul;21(7):1319-27. Carcinogenesis. 2000. PMID: 10874009
-
The signal pathways in azoxymethane-induced colon cancer and preventive implications.Cancer Biol Ther. 2009 Jul;8(14):1313-7. doi: 10.4161/cbt.8.14.8983. Epub 2009 Jul 11. Cancer Biol Ther. 2009. PMID: 19502780 Review.
-
Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents.Cancer Sci. 2004 Jun;95(6):475-80. doi: 10.1111/j.1349-7006.2004.tb03235.x. Cancer Sci. 2004. PMID: 15182426 Free PMC article. Review.
Cited by
-
Polyethylene glycol-mediated colorectal cancer chemoprevention: roles of epidermal growth factor receptor and Snail.Mol Cancer Ther. 2008 Sep;7(9):3103-11. doi: 10.1158/1535-7163.MCT-08-0434. Mol Cancer Ther. 2008. PMID: 18790788 Free PMC article.
-
Both stromal cell and colonocyte epidermal growth factor receptors control HCT116 colon cancer cell growth in tumor xenografts.Carcinogenesis. 2012 Oct;33(10):1930-9. doi: 10.1093/carcin/bgs231. Epub 2012 Jul 12. Carcinogenesis. 2012. PMID: 22791816 Free PMC article.
-
Epidermal growth factor receptor is required for colonic tumor promotion by dietary fat in the azoxymethane/dextran sulfate sodium model: roles of transforming growth factor-{alpha} and PTGS2.Clin Cancer Res. 2009 Nov 15;15(22):6780-9. doi: 10.1158/1078-0432.CCR-09-1678. Epub 2009 Nov 10. Clin Cancer Res. 2009. PMID: 19903783 Free PMC article.
-
PDGFA-associated protein 1 is a novel target of c-Myc and contributes to colorectal cancer initiation and progression.Cancer Commun (Lond). 2022 Aug;42(8):750-767. doi: 10.1002/cac2.12322. Epub 2022 Jun 18. Cancer Commun (Lond). 2022. PMID: 35716012 Free PMC article.
-
Animal models of colitis-associated carcinogenesis.J Biomed Biotechnol. 2011;2011:342637. doi: 10.1155/2011/342637. Epub 2011 Jan 12. J Biomed Biotechnol. 2011. PMID: 21274454 Free PMC article. Review.
References
-
- Vogelstein B, Kinzler K. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. - PubMed
-
- Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8:11–31. - PubMed
-
- Half E, Broaddus R, Danenberg KD, Danenberg PV, Ayers GD, Sinicrope FA. HER-2 receptor expression, localization, and activation in colorectal cancer cell lines and human tumors. Int J Cancer. 2004;108:540–548. - PubMed
-
- Maurer CA, Friess H, Kretschmann B, et al. Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol. 1998;29:771–777. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
