

Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance
- PMID: 17234885
- PMCID: PMC1770902
- DOI: 10.1101/gad.1505307
Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance
Abstract
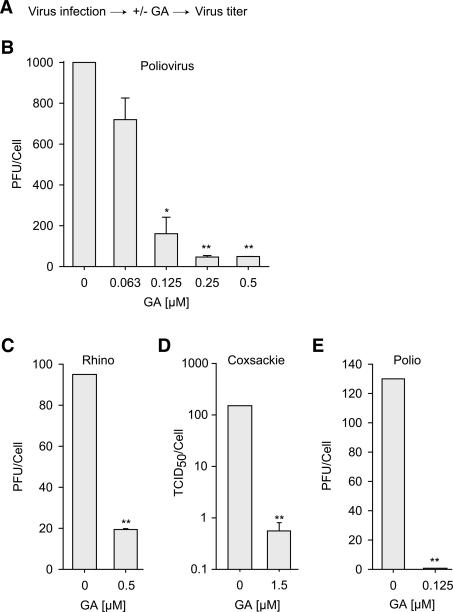
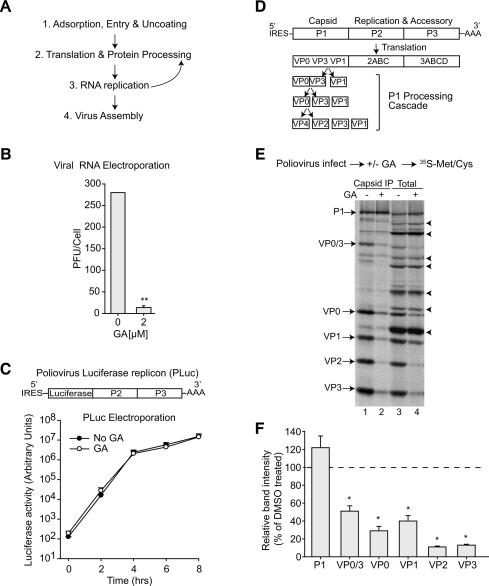
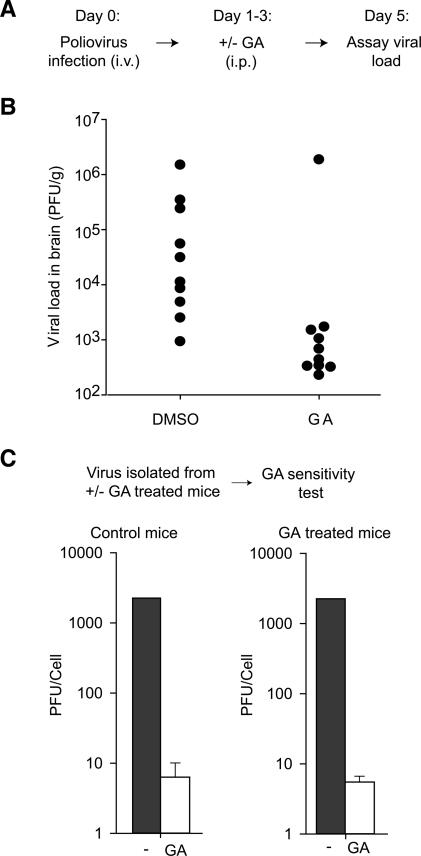
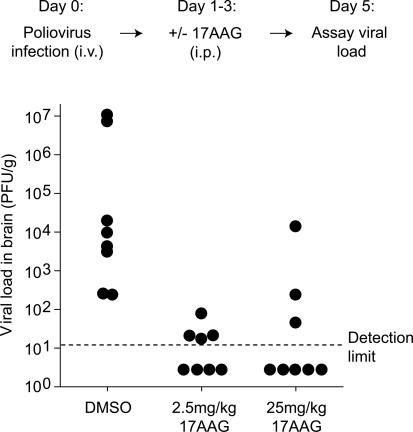
The genome diversity of RNA viruses allows for rapid adaptation to a wide variety of adverse conditions. Accordingly, viruses can escape inhibition by most antiviral compounds targeting either viral or host factors. Here we exploited the capacity of RNA viruses for rapid adaptation to explore the evolutionary constraints of chaperone-mediated protein folding. We hypothesized that inhibiting a host molecular chaperone required for folding of a viral protein would force the virus to evolve an alternate folding strategy. We identified the chaperone Hsp90 as an essential factor for folding and maturation of picornavirus capsid proteins. Pharmacological inhibition of Hsp90 impaired the replication of poliovirus, rhinovirus, and coxsackievirus in cell culture. Strikingly, anti-Hsp90 treatment did not yield drug-resistant viruses, suggesting that the complexity of capsid folding precludes the emergence of alternate folding pathways. These results reveal tight evolutionary constraints on chaperone-mediated protein folding, which may be exploited for viral inhibition in vivo. Indeed, Hsp90 inhibitors drastically reduced poliovirus replication in infected animals without the emergence of drug-resistant escape mutants. We propose that targeting folding of viral proteins may provide a general antiviral strategy that is refractory to development of drug resistance.
Figures


References
-
- Ali M.M., Roe S.M., Vaughan C.K., Meyer P., Panaretou B., Piper P.W., Prodromou C., Pearl L.H., Roe S.M., Vaughan C.K., Meyer P., Panaretou B., Piper P.W., Prodromou C., Pearl L.H., Vaughan C.K., Meyer P., Panaretou B., Piper P.W., Prodromou C., Pearl L.H., Meyer P., Panaretou B., Piper P.W., Prodromou C., Pearl L.H., Panaretou B., Piper P.W., Prodromou C., Pearl L.H., Piper P.W., Prodromou C., Pearl L.H., Prodromou C., Pearl L.H., Pearl L.H. Crystal structure of an Hsp90–nucleotide–p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. - PMC - PubMed
-
- Basavappa R., Syed R., Flore O., Icenogle J.P., Filman D.J., Hogle J.M., Syed R., Flore O., Icenogle J.P., Filman D.J., Hogle J.M., Flore O., Icenogle J.P., Filman D.J., Hogle J.M., Icenogle J.P., Filman D.J., Hogle J.M., Filman D.J., Hogle J.M., Hogle J.M. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: Structure of the empty capsid assembly intermediate at 2.9 Å resolution. Protein Sci. 1994;3:1651–1669. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources