

Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR
- PMID: 17235394
- PMCID: PMC1765518
- DOI: 10.1172/JCI30376
Direct interaction with filamins modulates the stability and plasma membrane expression of CFTR
Abstract
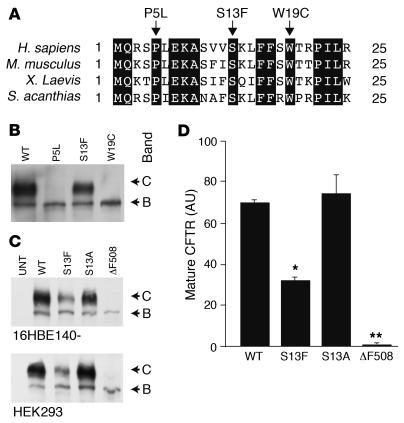
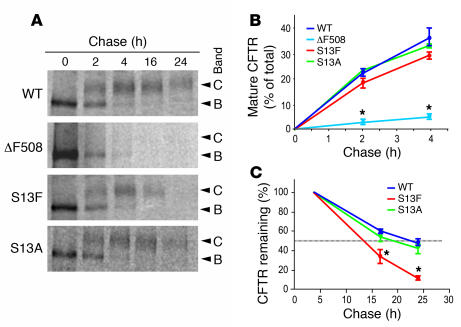
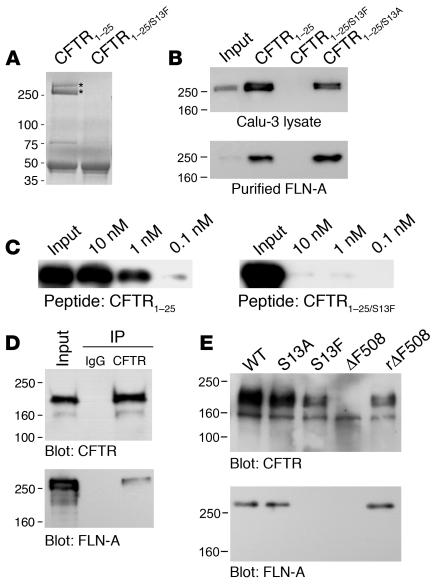
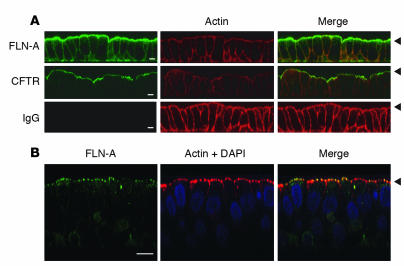
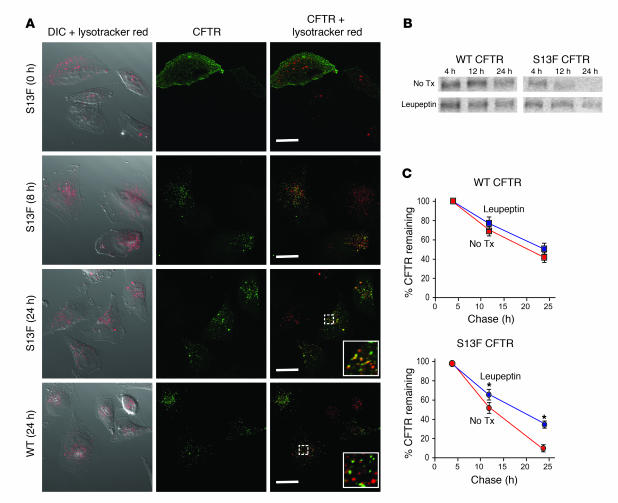
The role of the cystic fibrosis transmembrane conductance regulator (CFTR) as a cAMP-dependent chloride channel on the apical membrane of epithelia is well established. However, the processes by which CFTR is regulated on the cell surface are not clear. Here we report the identification of a protein-protein interaction between CFTR and the cytoskeletal filamin proteins. Using proteomic approaches, we identified filamins as proteins that associate with the extreme CFTR N terminus. Furthermore, we identified a disease-causing missense mutation in CFTR, serine 13 to phenylalanine (S13F), which disrupted this interaction. In cells, filamins tethered plasma membrane CFTR to the underlying actin network. This interaction stabilized CFTR at the cell surface and regulated the plasma membrane dynamics and confinement of the channel. In the absence of filamin binding, CFTR was internalized from the cell surface, where it prematurely accumulated in lysosomes and was ultimately degraded. Our data demonstrate what we believe to be a previously unrecognized role for the CFTR N terminus in the regulation of the plasma membrane stability and metabolic stability of CFTR. In addition, we elucidate the molecular defect associated with the S13F mutation.
Figures
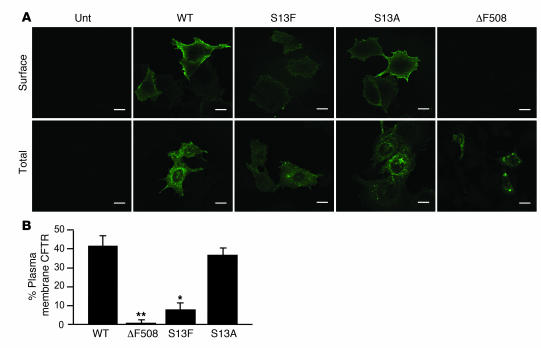
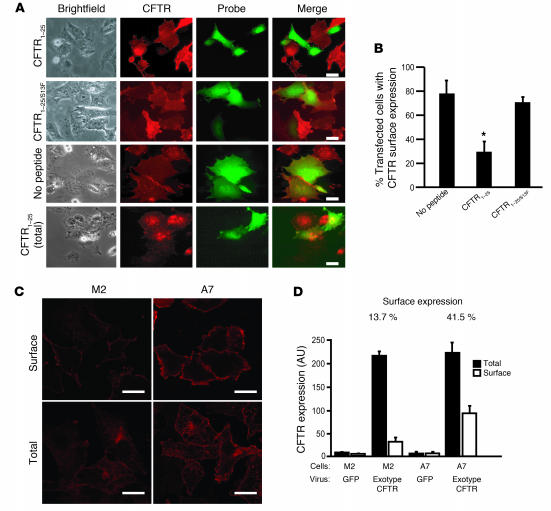
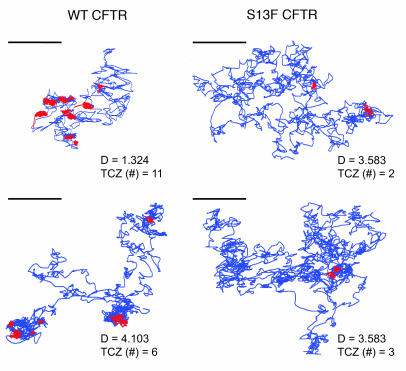
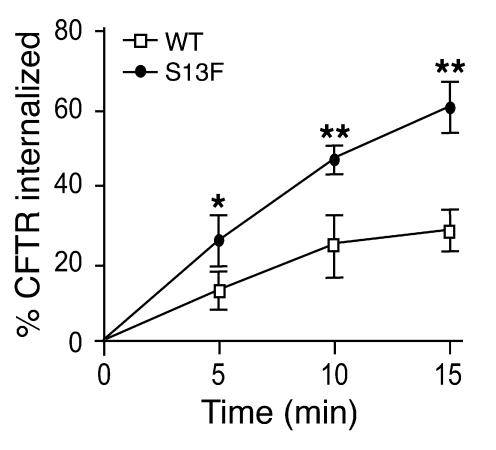
References
-
- Welsh M.J., Smith A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. - PubMed
-
- Zielenski J., Tsui L.C. Cystic fibrosis: genotypic and phenotypic variations. Annu. Rev. Genet. 1995;29:777–807. - PubMed
-
- Haardt M., Benharouga M., Lechardeur D., Kartner N., Lukacs G.L. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J. Biol. Chem. 1999;274:21873–21877. - PubMed
-
- Denning G.M., et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. - PubMed
-
- Du K., Sharma M., Lukacs G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005;12:17–25. - PubMed
