

PTEN-deficient intestinal stem cells initiate intestinal polyposis
- PMID: 17237784
- PMCID: PMC4681524
- DOI: 10.1038/ng1928
PTEN-deficient intestinal stem cells initiate intestinal polyposis
Abstract
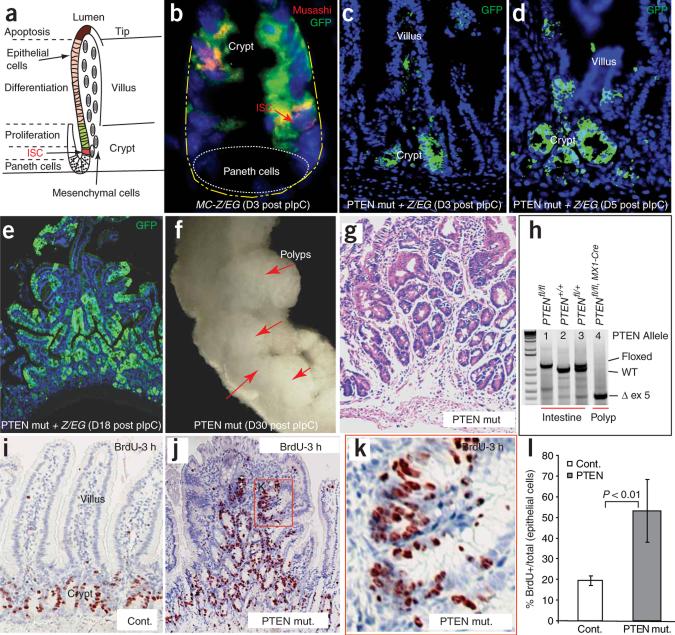
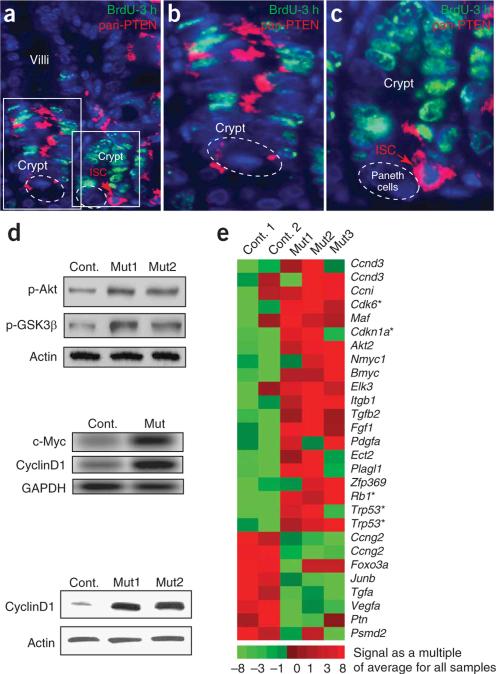
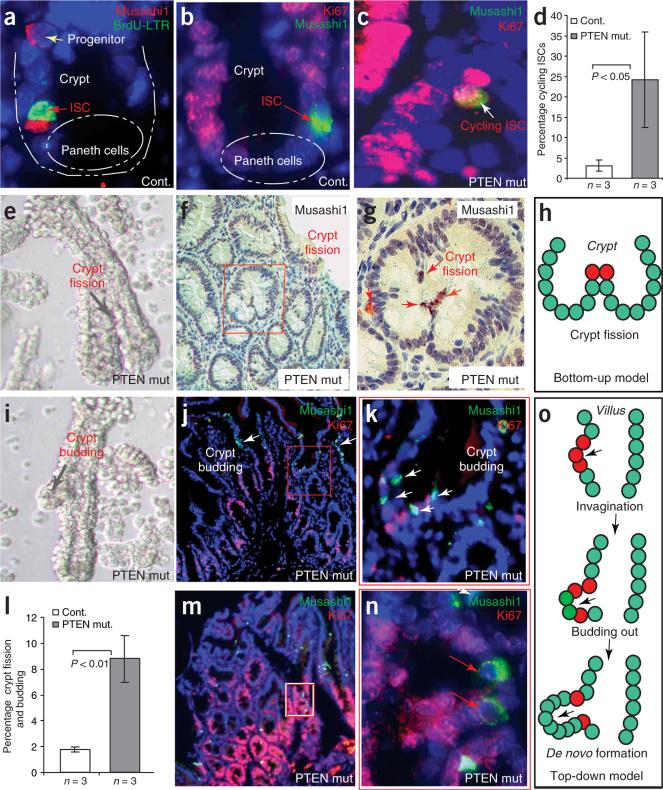
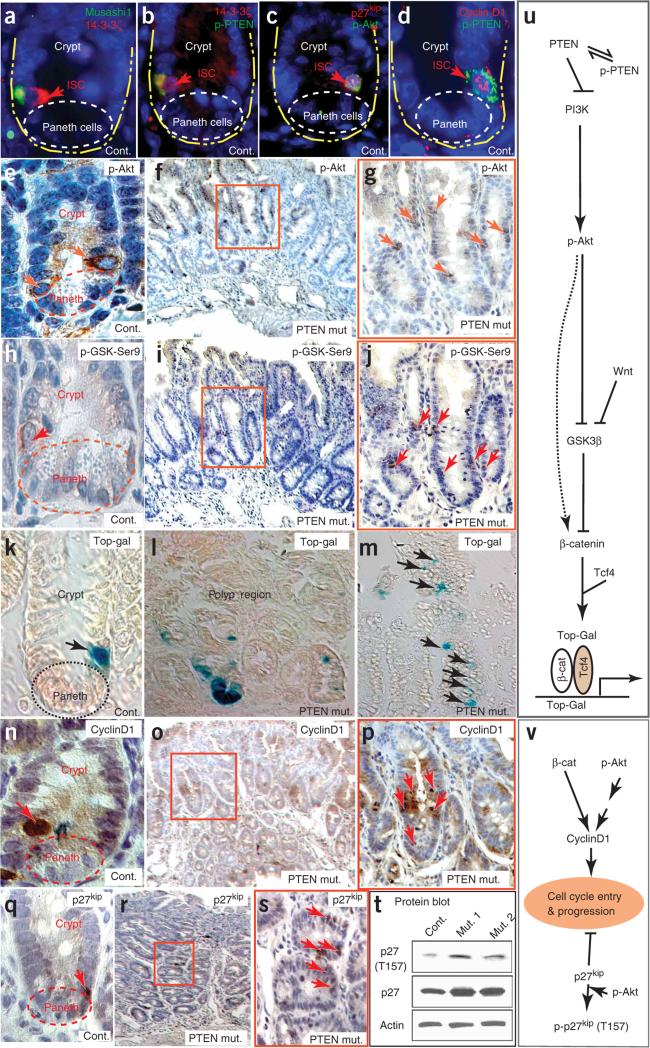
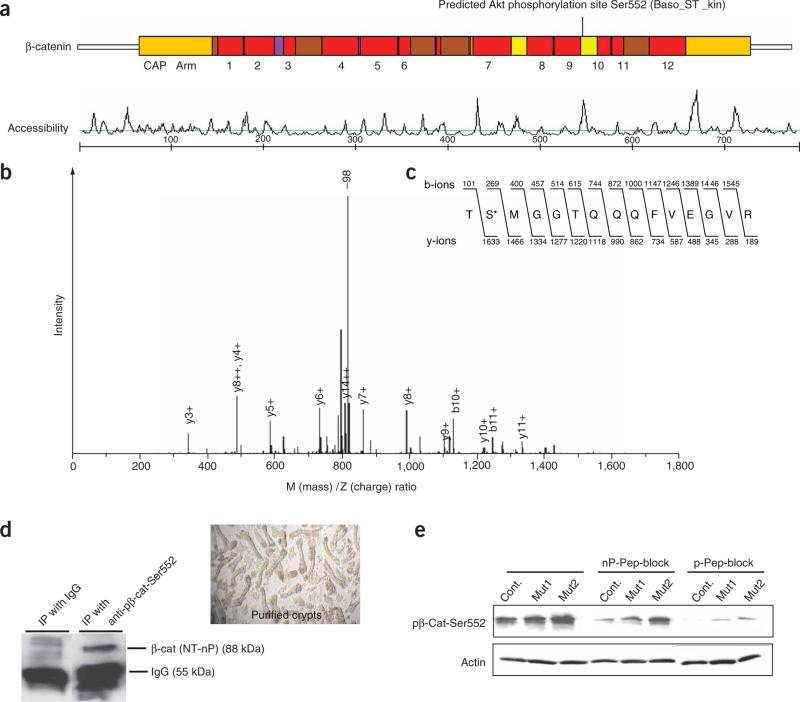
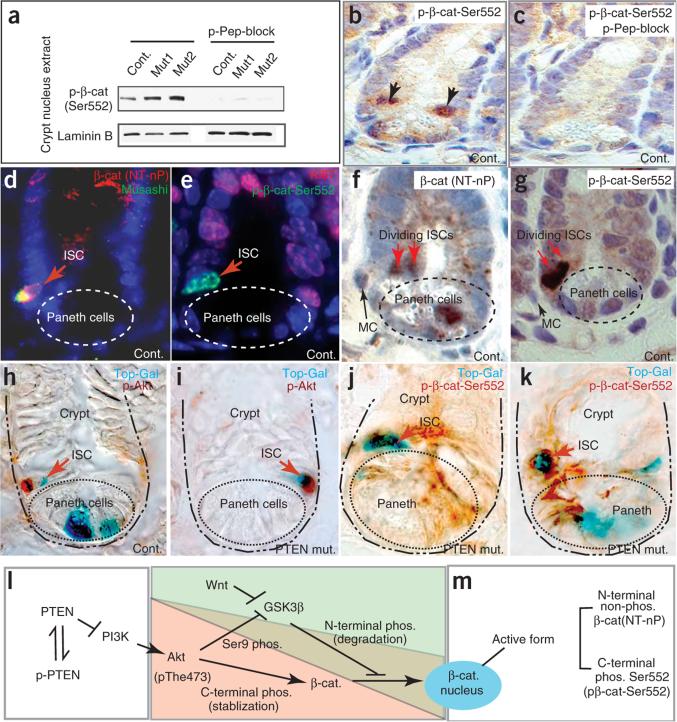
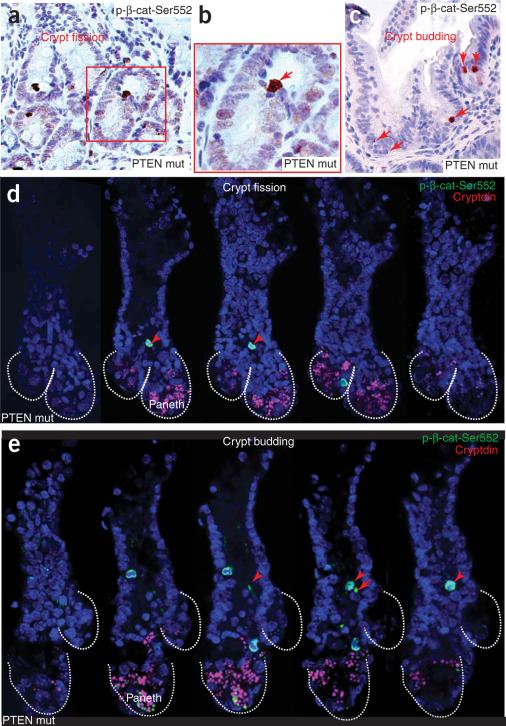
Intestinal polyposis, a precancerous neoplasia, results primarily from an abnormal increase in the number of crypts, which contain intestinal stem cells (ISCs). In mice, widespread deletion of the tumor suppressor Phosphatase and tensin homolog (PTEN) generates hamartomatous intestinal polyps with epithelial and stromal involvement. Using this model, we have established the relationship between stem cells and polyp and tumor formation. PTEN helps govern the proliferation rate and number of ISCs and loss of PTEN results in an excess of ISCs. In PTEN-deficient mice, excess ISCs initiate de novo crypt formation and crypt fission, recapitulating crypt production in fetal and neonatal intestine. The PTEN-Akt pathway probably governs stem cell activation by helping control nuclear localization of the Wnt pathway effector beta-catenin. Akt phosphorylates beta-catenin at Ser552, resulting in a nuclear-localized form in ISCs. Our observations show that intestinal polyposis is initiated by PTEN-deficient ISCs that undergo excessive proliferation driven by Akt activation and nuclear localization of beta-catenin.
Figures
References
-
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. - PubMed
-
- Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. - PubMed
-
- Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. - PubMed
-
- Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J. Cell Sci. 2002;115:2381–2388. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
