

Reductive genome evolution from the mother of Rickettsia
- PMID: 17238289
- PMCID: PMC1779305
- DOI: 10.1371/journal.pgen.0030014
Reductive genome evolution from the mother of Rickettsia
Abstract
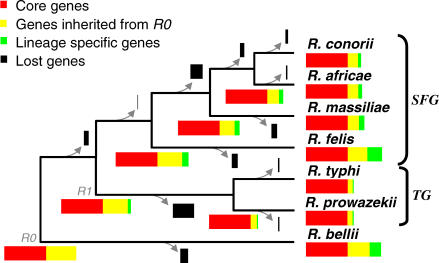
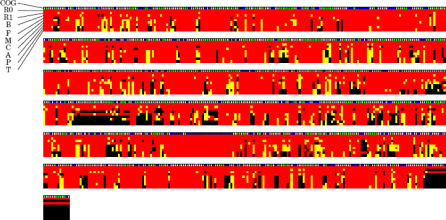
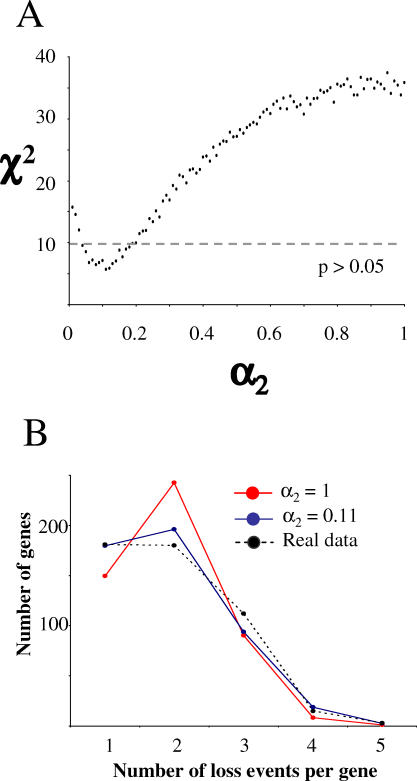
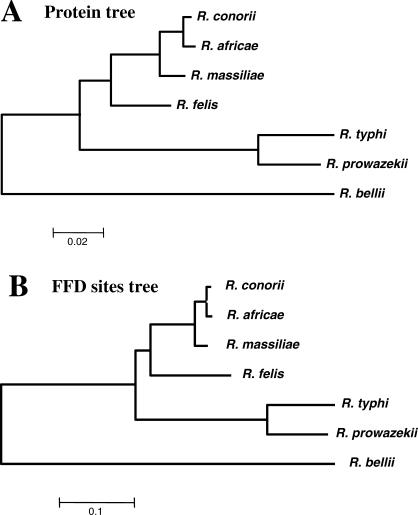
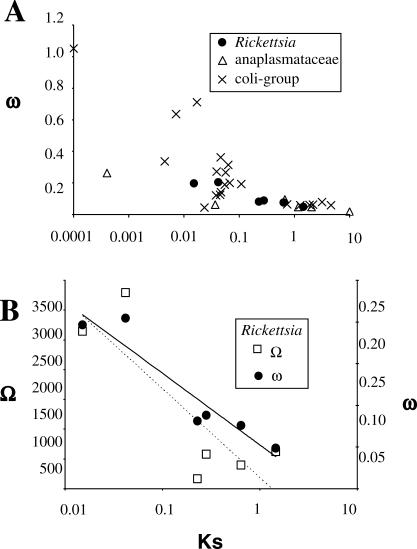
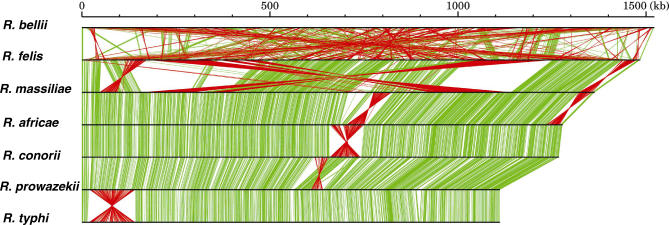
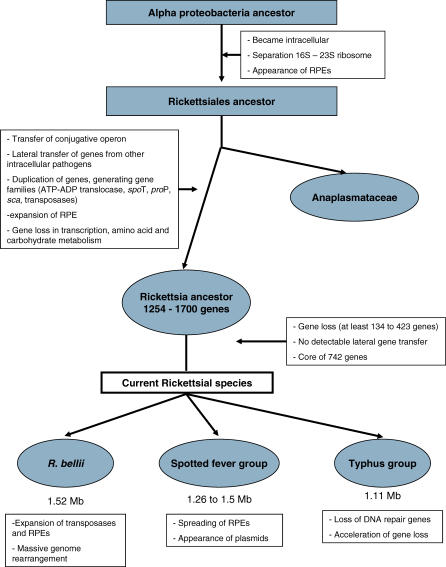
The Rickettsia genus is a group of obligate intracellular alpha-proteobacteria representing a paradigm of reductive evolution. Here, we investigate the evolutionary processes that shaped the genomes of the genus. The reconstruction of ancestral genomes indicates that their last common ancestor contained more genes, but already possessed most traits associated with cellular parasitism. The differences in gene repertoires across modern Rickettsia are mainly the result of differential gene losses from the ancestor. We demonstrate using computer simulation that the propensity of loss was variable across genes during this process. We also analyzed the ratio of nonsynonymous to synonymous changes (Ka/Ks) calculated as an average over large sets of genes to assay the strength of selection acting on the genomes of Rickettsia, Anaplasmataceae, and free-living gamma-proteobacteria. As a general trend, Ka/Ks were found to decrease with increasing divergence between genomes. The high Ka/Ks for closely related genomes are probably due to a lag in the removal of slightly deleterious nonsynonymous mutations by natural selection. Interestingly, we also observed a decrease of the rate of gene loss with increasing divergence, suggesting a similar lag in the removal of slightly deleterious pseudogene alleles. For larger divergence (Ks > 0.2), Ka/Ks converge toward similar values indicating that the levels of selection are roughly equivalent between intracellular alpha-proteobacteria and their free-living relatives. This contrasts with the view that obligate endocellular microorganisms tend to evolve faster as a consequence of reduced effectiveness of selection, and suggests a major role of enhanced background mutation rates on the fast protein divergence in the obligate intracellular alpha-proteobacteria.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
References
-
- Wernegreen JJ. For better or worse: Genomic consequences of intracellular mutualism and parasitism. Curr Opin Genet Dev. 2005;15:572–583. - PubMed
-
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. - PubMed
-
- Ogata H, Audic S, Renesto-Audiffren P, Fournier PE, Barbe V, et al. Mechanisms of evolution in Rickettsia conorii and R. prowazekii . Science. 2001;293:2093–2098. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
