

PILER-CR: fast and accurate identification of CRISPR repeats
- PMID: 17239253
- PMCID: PMC1790904
- DOI: 10.1186/1471-2105-8-18
PILER-CR: fast and accurate identification of CRISPR repeats
Abstract
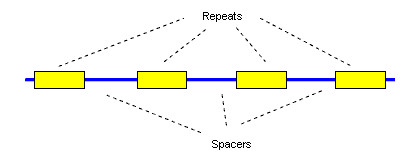
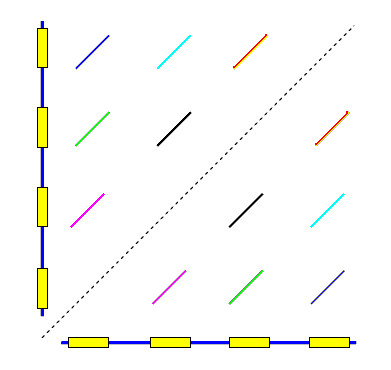
Background: Sequencing of prokaryotic genomes has recently revealed the presence of CRISPR elements: short, highly conserved repeats separated by unique sequences of similar length. The distinctive sequence signature of CRISPR repeats can be found using general-purpose repeat- or pattern-finding software tools. However, the output of such tools is not always ideal for studying these repeats, and significant effort is sometimes needed to build additional tools and perform manual analysis of the output.
Results: We present PILER-CR, a program specifically designed for the identification and analysis of CRISPR repeats. The program executes rapidly, completing a 5 Mb genome in around 5 seconds on a current desktop computer. We validate the algorithm by manual curation and by comparison with published surveys of these repeats, finding that PILER-CR has both high sensitivity and high specificity. We also present a catalogue of putative CRISPR repeats identified in a comprehensive analysis of 346 prokaryotic genomes.
Conclusion: PILER-CR is a useful tool for rapid identification and classification of CRISPR repeats. The software is donated to the public domain. Source code and a Linux binary are freely available at http://www.drive5.com/pilercr.
Figures
References
-
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghagen NS, Venter JC. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
