

Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis
- PMID: 17241866
- PMCID: PMC2190109
- DOI: 10.1053/j.gastro.2006.10.034
Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis
Abstract
Background & aims: Inherited syndromes of intrahepatic cholestasis commonly result from mutations in the genes SERPINA1 (alpha(1)-antitrypsin deficiency), JAG1 (Alagille syndrome), ATP8B1 (progressive familial intrahepatic cholestasis type 1 [PFIC1]), ABCB11 (PFIC2), and ABCB4 (PFIC3). However, the large gene sizes and lack of mutational hotspots make it difficult to survey for disease-causing mutations in clinical practice. Here, we aimed to develop a technological tool that reads out the nucleotide sequence of these genes rapidly and accurately.
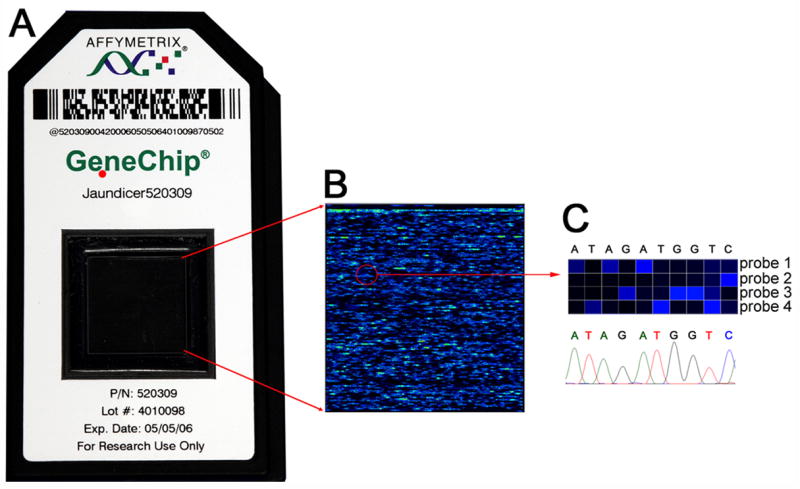
Methods: 25-mer nucleotide probes were designed to identify each base for all exons, 10 bases of intronic sequence bordering exons, 280-500 bases upstream from the first exon for each gene, and 350 bases of the second intron of the JAG1 gene and tiled using the Affymetrix resequencing platform. We then developed high-fidelity polymerase chain reactions to produce amplicons using 1 mL of blood from each subject; amplicons were hybridized to the chip, and nucleotide calls were validated by standard capillary sequencing methods.
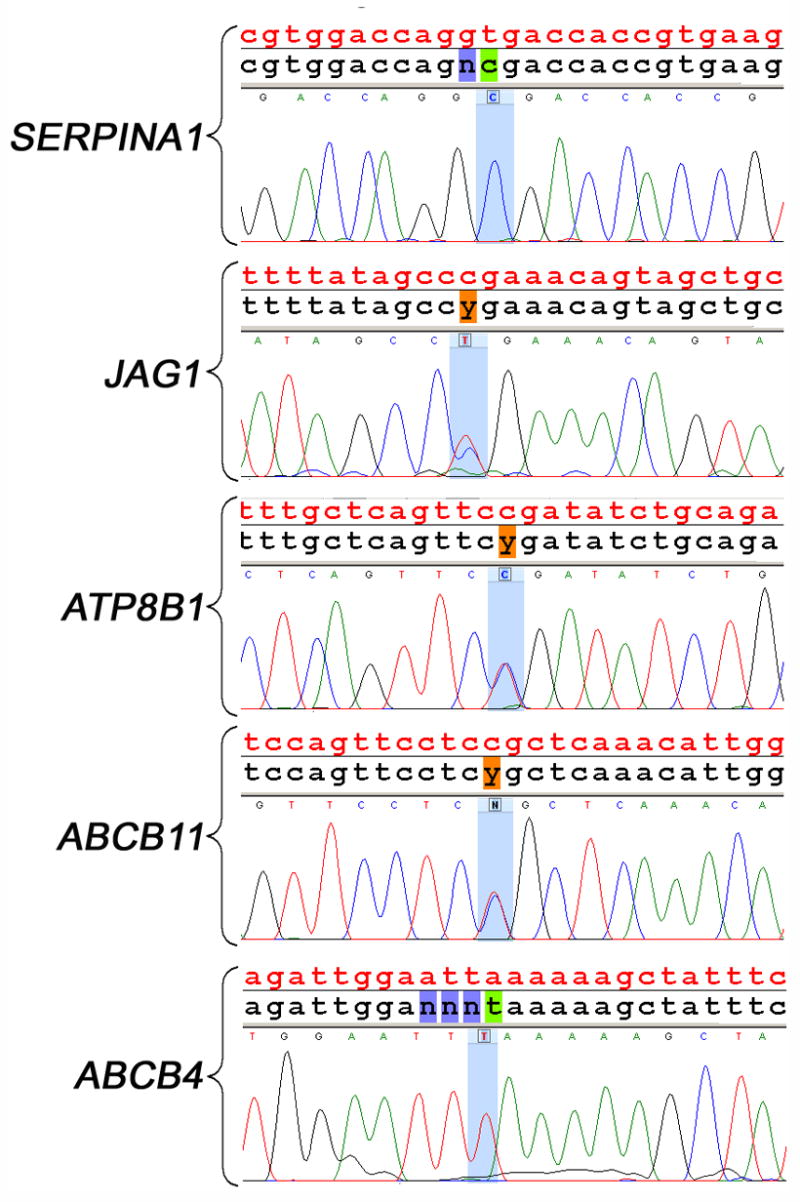
Results: Hybridization of amplicons with the chip produced a high nucleotide sequence readout for all 5 genes in a single assay, with an automated call rate of 93.5% (range, 90.3%-95.7%). The accuracy of nucleotide calls was 99.99% when compared with capillary sequencing. Testing the chip on subjects with cholestatic syndromes identified disease-causing mutations in SERPINA1, JAG1, ATP8B1, ABCB11, or ABCB4.
Conclusions: The resequencing chip efficiently reads SERPINA1, JAG1, ATP8B1, ABCB11, and ABCB4 with a high call rate and accuracy in one assay and identifies disease-causing mutations.
Figures
References
-
- Balistreri WF, Bezerra JA. Whatever happened to "neonatal hepatitis"? Clin Liver Dis. 2006;10:27–53. v. - PubMed
-
- Sharp HL, Bridges RA, Krivit W, Freier EF. Cirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorder. J Lab Clin Med. 1969;73:934–9. - PubMed
-
- Odievre M, Martin JP, Hadchouel M, Alagille D. Alpha1-antitrypsin deficiency and liver disease in children: phenotypes, manifestations, and prognosis. Pediatrics. 1976;57:226–31. - PubMed
-
- Balistreri WF, Bezerra JA, Jansen P, Karpen SJ, Shneider BL, Suchy FJ. Intrahepatic cholestasis: summary of an American Association for the Study of Liver Diseases single-topic conference. Hepatology. 2005;42:222–35. - PubMed
-
- Alagille D, Estrada A, Hadchouel M, Gautier M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
