

Comparative molecular dynamics analysis of tapasin-dependent and -independent MHC class I alleles
- PMID: 17242432
- PMCID: PMC2203297
- DOI: 10.1110/ps.062568407
Comparative molecular dynamics analysis of tapasin-dependent and -independent MHC class I alleles
Abstract
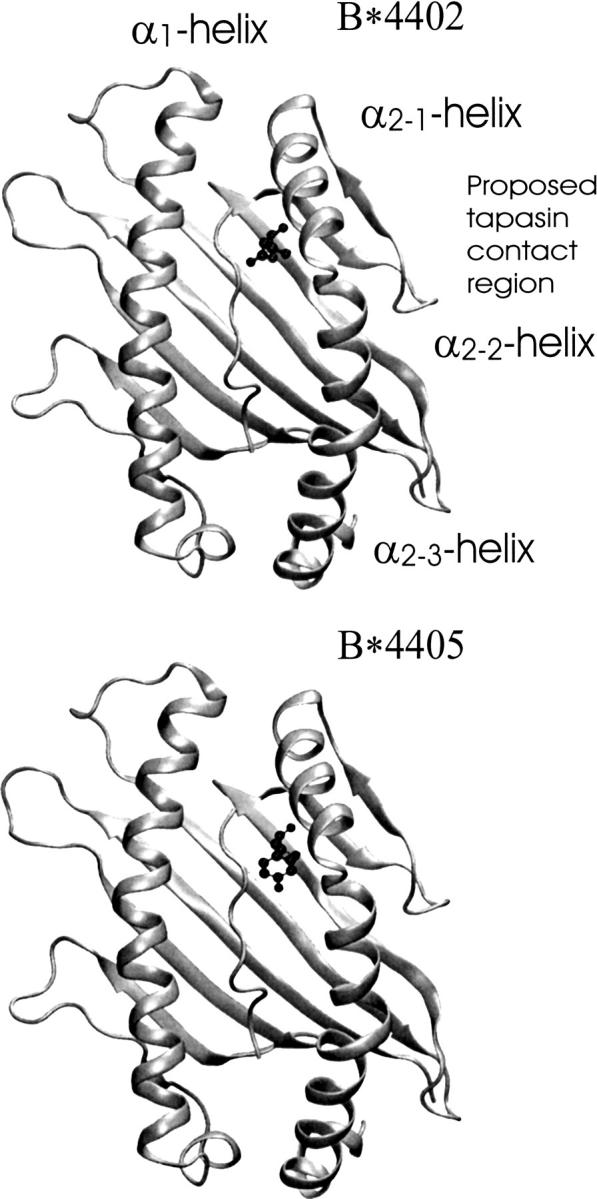
MHC class I molecules load antigenic peptides in the endoplasmic reticulum and present them at the cell surface. Efficiency of peptide loading depends on the class I allele and can involve interaction with tapasin and other proteins of the loading complex. Allele HLA-B*4402 (Asp at position 116) depends on tapasin for efficient peptide loading, whereas HLA-B*4405 (identical to B*4402 except for Tyr116) can efficiently load peptides in the absence of tapasin. Both alleles adopt very similar structures in the presence of the same peptide. Comparative unrestrained molecular dynamics simulations on the alpha(1)/alpha(2) peptide binding domains performed in the presence of bound peptides resulted in structures in close agreement with experiments for both alleles. In the absence of peptides, allele-specific conformational changes occurred in the first segment of the alpha(2)-helix that flanks the peptide C-terminal binding region (F-pocket) and contacts residue 116. This segment is also close to the proposed tapasin contact region. For B*4402, a shift toward an altered F-pocket structure deviating significantly from the bound form was observed. Subsequent free energy simulations on induced F-pocket opening in B*4402 confirmed a conformation that deviated significantly from the bound structure. For B*4405, a free energy minimum close to the bound structure was found. The simulations suggest that B*4405 has a greater tendency to adopt a peptide receptive conformation in the absence of peptide, allowing tapasin-independent peptide loading. A possible role of tapasin could be the stabilization of a peptide-receptive class I conformation for HLA-B*4402 and other tapasin-dependent alleles.
Figures






References
-
- Bouvier, M. 2003. Accessory proteins and the assembly of human class I MHC molecules: A molecular and structural perspective. Mol. Immunol. 39: 697–706. - PubMed
-
- Bouvier, M. and Wiley, D.C. 1998. Structural characterization of a soluble and partial folded class I major histocompatibility chain/β2m heterodimer. Nat. Struct. Biol. 5: 377–384. - PubMed
-
- Case, D.A., Darden, T.A., Cheatham III, T.E., Simmerling, C.L., Wang, J., Duke, R.E., Luo, R., Merz, K.M., Wang, B., and Pearlman, D.A., et al. 2004. AMBER8. University of California, San Francisco.
-
- Darden, T., York, D., and Pedersen, L. 1993. Particle mesh Ewald: An NlogN method for Ewald sums in large systems. J. Chem. Phys. 98: 10089–10092.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
