

Metabolic catastrophe as a means to cancer cell death
- PMID: 17251378
- PMCID: PMC2857576
- DOI: 10.1242/jcs.03349
Metabolic catastrophe as a means to cancer cell death
Abstract
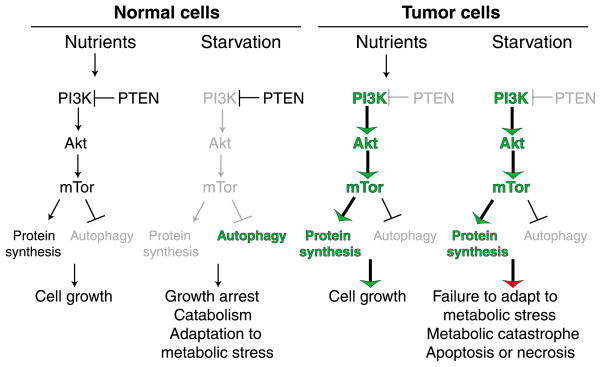
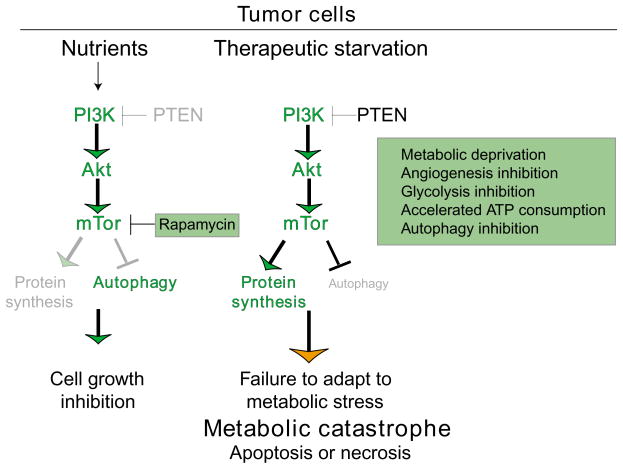
During tumorigenesis, normal growth mechanisms are deregulated and safeguards that eliminate abnormal cells by apoptosis are disabled. Tumor cells must also increase nutrient uptake and angiogenesis to support the upregulation of metabolism necessary for unrestricted growth. In addition, they have to rely on inefficient energy production by glycolysis. This glycolytic state can result from mutations that promote cell proliferation, the hypoxic tumor microenvironment and perhaps mitochondrial malfunction. Moreover, the very signals that enable unrestricted cell proliferation inhibit autophagy, which normally sustains cells during nutrient limitation. In tumors, inactivation of the autophagy pathway may enhance necrosis and inflammation and promote genomic instability, which can further enhance tumor growth. Thus, tumor cells cannot adapt efficiently to metabolic stress and could be induced to die by metabolic catastrophe, in which high energy demand is contrasted by insufficient energy production. Efforts to exploit this unique metabolic state clinically previously focused mainly on detecting tissue displaying increased glycolytic metabolism. The challenge now is to induce metabolic catastrophe therapeutically as an approach to killing the unkillable cells.
Figures

References
-
- Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. - PubMed
-
- Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. - PubMed
-
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. - PubMed
-
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
