

Splice site identification using probabilistic parameters and SVM classification
- PMID: 17254299
- PMCID: PMC1764471
- DOI: 10.1186/1471-2105-7-S5-S15
Splice site identification using probabilistic parameters and SVM classification
Erratum in
- BMC Bioinformatics. 2007 Jul 5;8:241
Abstract
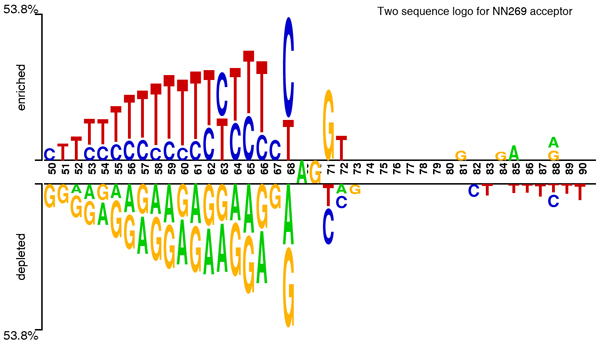
Background: Recent advances and automation in DNA sequencing technology has created a vast amount of DNA sequence data. This increasing growth of sequence data demands better and efficient analysis methods. Identifying genes in this newly accumulated data is an important issue in bioinformatics, and it requires the prediction of the complete gene structure. Accurate identification of splice sites in DNA sequences plays one of the central roles of gene structural prediction in eukaryotes. Effective detection of splice sites requires the knowledge of characteristics, dependencies, and relationship of nucleotides in the splice site surrounding region. A higher-order Markov model is generally regarded as a useful technique for modeling higher-order dependencies. However, their implementation requires estimating a large number of parameters, which is computationally expensive.
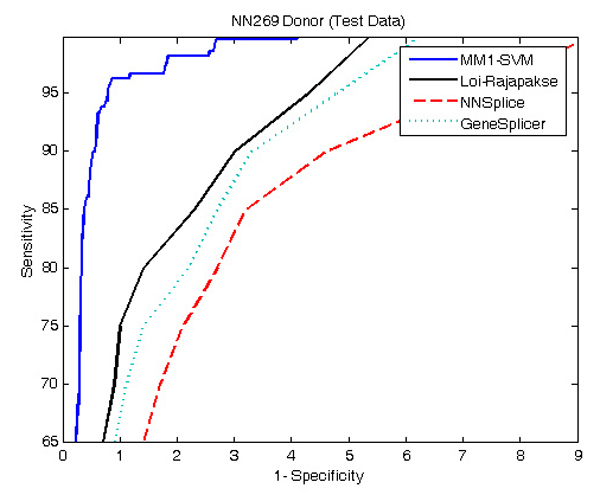
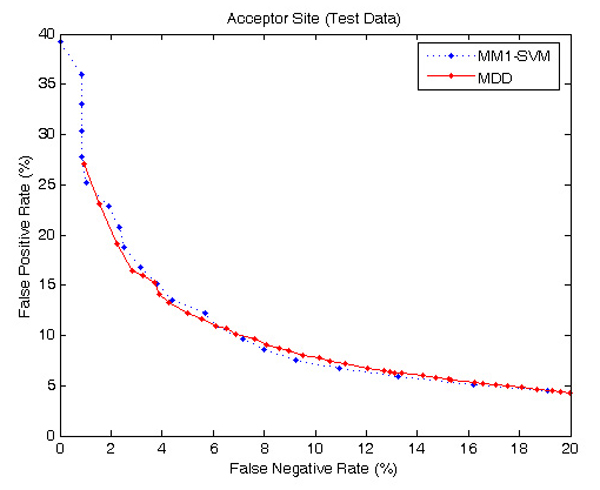
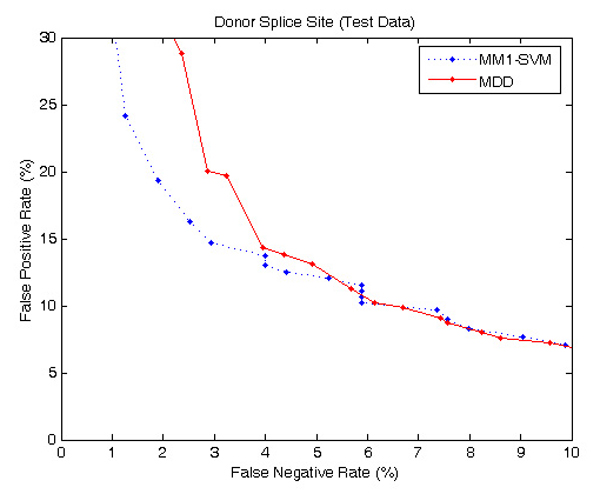
Results: The proposed method for splice site detection consists of two stages: a first order Markov model (MM1) is used in the first stage and a support vector machine (SVM) with polynomial kernel is used in the second stage. The MM1 serves as a pre-processing step for the SVM and takes DNA sequences as its input. It models the compositional features and dependencies of nucleotides in terms of probabilistic parameters around splice site regions. The probabilistic parameters are then fed into the SVM, which combines them nonlinearly to predict splice sites. When the proposed MM1-SVM model is compared with other existing standard splice site detection methods, it shows a superior performance in all the cases.
Conclusion: We proposed an effective pre-processing scheme for the SVM and applied it for the identification of splice sites. This is a simple yet effective splice site detection method, which shows a better classification accuracy and computational speed than some other more complex methods.
Figures
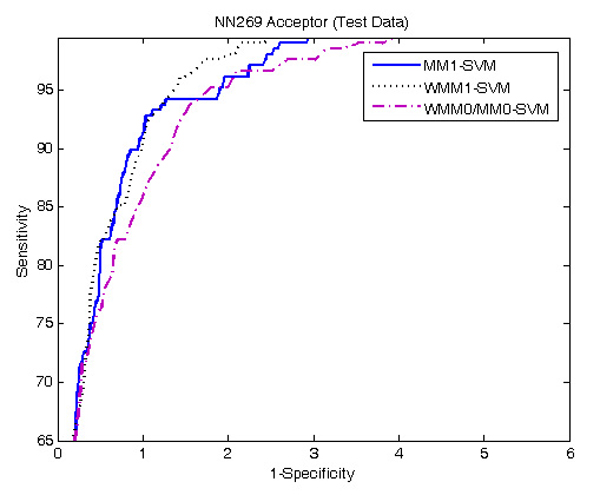
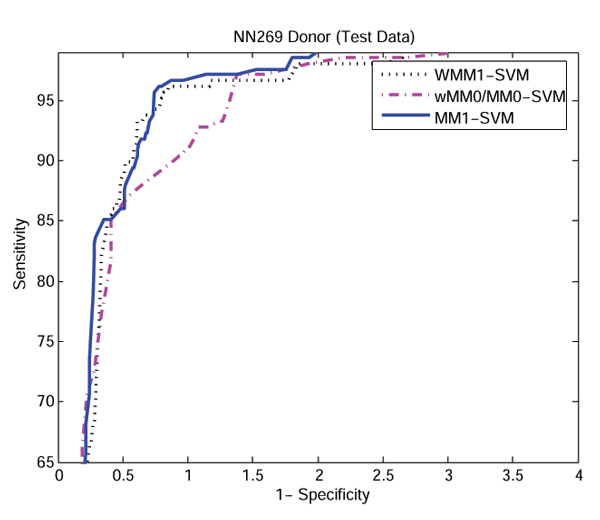
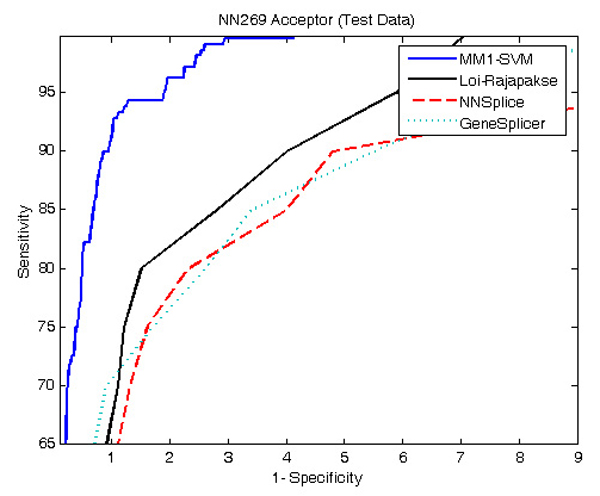

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
