

Refinement and expansion of signaling pathways: the osmotic response network in yeast
- PMID: 17267811
- PMCID: PMC1800927
- DOI: 10.1101/gr.5750507
Refinement and expansion of signaling pathways: the osmotic response network in yeast
Abstract
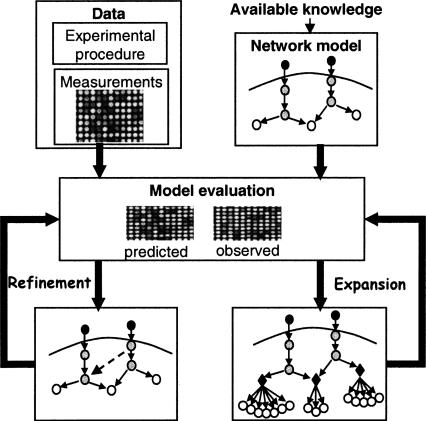
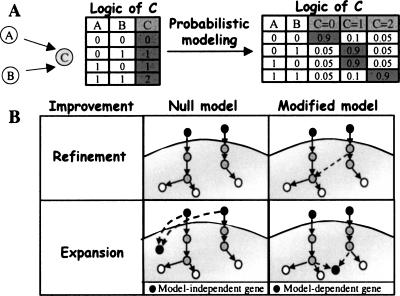
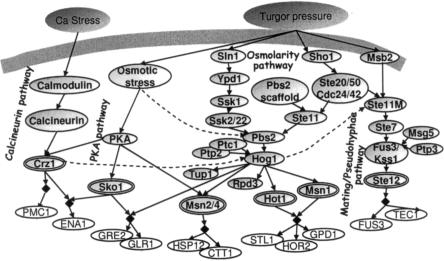
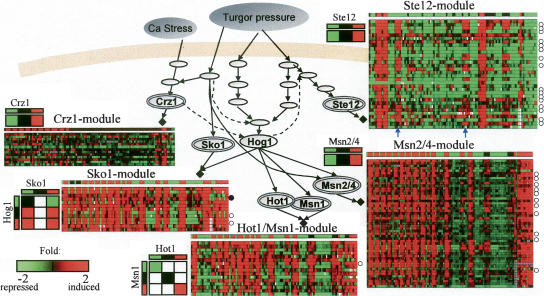
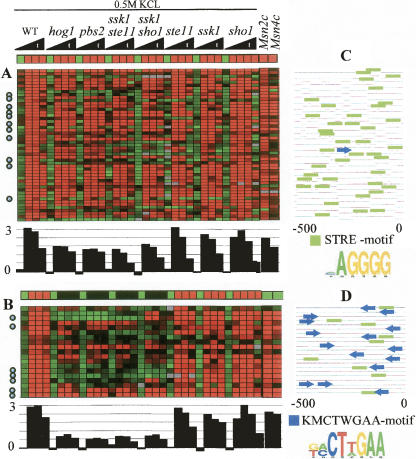
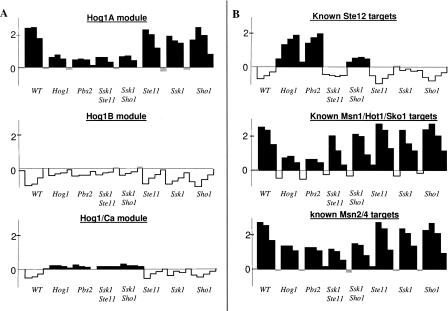
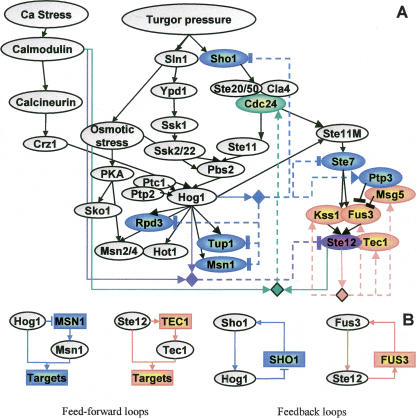
The analysis of large-scale genome-wide experiments carries the promise of dramatically broadening our understanding on biological networks. The challenge of systematic integration of experimental results with established biological knowledge on a pathway is still unanswered. Here we present a methodology that attempts to answer this challenge when investigating signaling pathways. We formalize existing qualitative knowledge as a probabilistic model that depicts known interactions between molecules (genes, proteins, etc.) as a network and known regulatory relations as logics. We present algorithms that analyze experimental results (e.g., transcription profiles) vis-à-vis the model and propose improvements to the model based on the fit to the experimental data. These algorithms refine the relations between model components, as well as expand the model to include new components that are regulated by components of the original network. Using our methodology, we have modeled together the knowledge on four established signaling pathways related to osmotic shock response in Saccharomyces cerevisiae. Using over 100 published transcription profiles, our refinement methodology revealed three cross talks in the network. The expansion procedure identified with high confidence large groups of genes that are coregulated by transcription factors from the original network via a common logic. The results reveal a novel delicate repressive effect of the HOG pathway on many transcriptional target genes and suggest an unexpected alternative functional mode of the MAP kinase Hog1. These results demonstrate that, by integrated analysis of data and of well-defined knowledge, one can generate concrete biological hypotheses about signaling cascades and their downstream regulatory programs.
Figures
Similar articles
-
A quantitative study of the Hog1 MAPK response to fluctuating osmotic stress in Saccharomyces cerevisiae.PLoS One. 2010 Mar 4;5(3):e9522. doi: 10.1371/journal.pone.0009522. PLoS One. 2010. PMID: 20209100 Free PMC article.
-
Transcriptional regulatory networks in Saccharomyces cerevisiae.Science. 2002 Oct 25;298(5594):799-804. doi: 10.1126/science.1075090. Science. 2002. PMID: 12399584
-
Genomewide identification of Sko1 target promoters reveals a regulatory network that operates in response to osmotic stress in Saccharomyces cerevisiae.Eukaryot Cell. 2005 Aug;4(8):1343-52. doi: 10.1128/EC.4.8.1343-1352.2005. Eukaryot Cell. 2005. PMID: 16087739 Free PMC article.
-
Toward a genomic view of the gene expression program regulated by osmostress in yeast.OMICS. 2010 Dec;14(6):619-27. doi: 10.1089/omi.2010.0046. Epub 2010 Aug 20. OMICS. 2010. PMID: 20726780 Review.
-
Yeast osmoregulation.Methods Enzymol. 2007;428:29-45. doi: 10.1016/S0076-6879(07)28002-4. Methods Enzymol. 2007. PMID: 17875410 Review.
Cited by
-
Applications of different weighting schemes to improve pathway-based analysis.Comp Funct Genomics. 2011;2011:463645. doi: 10.1155/2011/463645. Epub 2011 May 22. Comp Funct Genomics. 2011. PMID: 21687588 Free PMC article.
-
Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction.Mol Syst Biol. 2009;5:331. doi: 10.1038/msb.2009.87. Epub 2009 Dec 1. Mol Syst Biol. 2009. PMID: 19953085 Free PMC article.
-
REFINING CELLULAR PATHWAY MODELS USING AN ENSEMBLE OF HETEROGENEOUS DATA SOURCES.Ann Appl Stat. 2018 Sep;12(3):1361-1384. doi: 10.1214/16-aoas915. Epub 2018 Sep 11. Ann Appl Stat. 2018. PMID: 36506698 Free PMC article.
-
A network-based approach for predicting missing pathway interactions.PLoS Comput Biol. 2012;8(8):e1002640. doi: 10.1371/journal.pcbi.1002640. Epub 2012 Aug 16. PLoS Comput Biol. 2012. PMID: 22916002 Free PMC article.
-
Environmental stresses and clinical drugs paralyze a cell.Commun Integr Biol. 2009 May;2(3):275-8. doi: 10.4161/cib.2.3.8226. Commun Integr Biol. 2009. PMID: 19641750 Free PMC article.
References
-
- Bar-Joseph Z., Gerber G.K., Lee T.I., Rinaldi N.J., Yoo J.Y., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Gerber G.K., Lee T.I., Rinaldi N.J., Yoo J.Y., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Lee T.I., Rinaldi N.J., Yoo J.Y., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Rinaldi N.J., Yoo J.Y., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Yoo J.Y., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Robert F., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Gordon D.B., Fraenkel E., Jaakkola T.S., Young R.A., Fraenkel E., Jaakkola T.S., Young R.A., Jaakkola T.S., Young R.A., Young R.A., et al. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 2003;21:1337–1342. - PubMed
-
- Beer M.A., Tavazoie S., Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. - PubMed
-
- Calvano S.E., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Xiao W., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Richards D.R., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Felciano R.M., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Baker H.V., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Cho R.J., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Chen R.O., Brownstein B.H., Cobb J.P., Tschoeke S.K., Brownstein B.H., Cobb J.P., Tschoeke S.K., Cobb J.P., Tschoeke S.K., Tschoeke S.K., et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. - PubMed
-
- Covert M.W., Knight E.M., Reed J.L., Herrgard M.J., Palsson B.O., Knight E.M., Reed J.L., Herrgard M.J., Palsson B.O., Reed J.L., Herrgard M.J., Palsson B.O., Herrgard M.J., Palsson B.O., Palsson B.O. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. - PubMed
-
- De Nadal E., Zapater M., Alepuz P.M., Sumoy L., Mas G., Posas F., Zapater M., Alepuz P.M., Sumoy L., Mas G., Posas F., Alepuz P.M., Sumoy L., Mas G., Posas F., Sumoy L., Mas G., Posas F., Mas G., Posas F., Posas F. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature. 2004;427:370–374. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases