

Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32-33
- PMID: 17273976
- PMCID: PMC1821099
- DOI: 10.1086/512248
Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32-33
Abstract
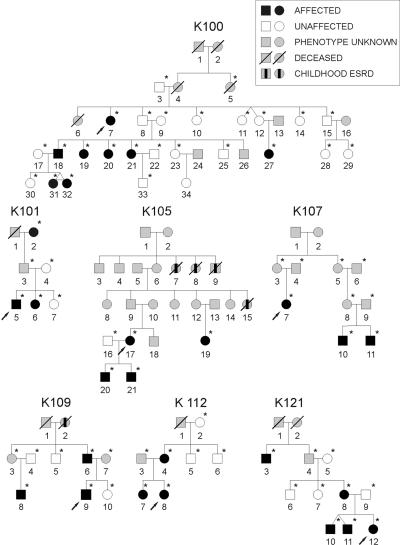
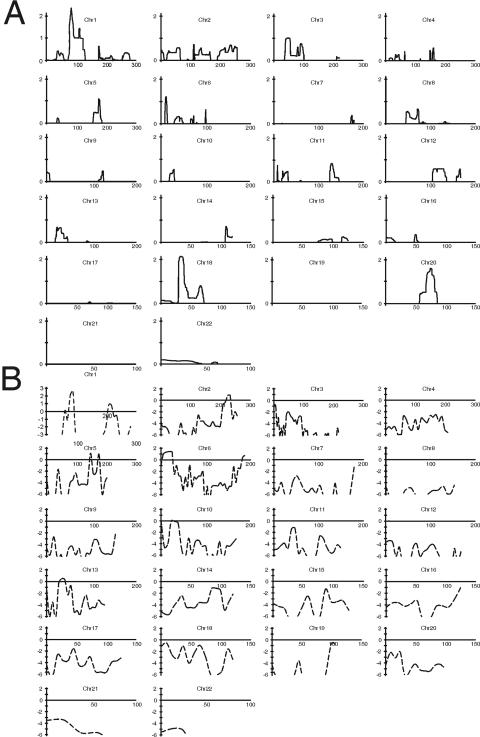
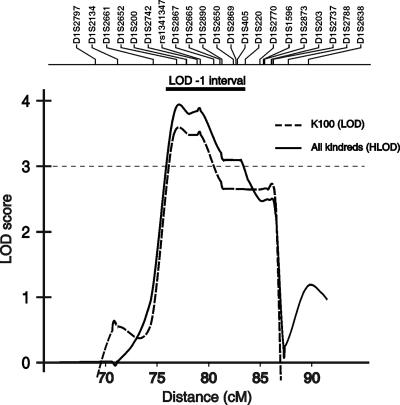
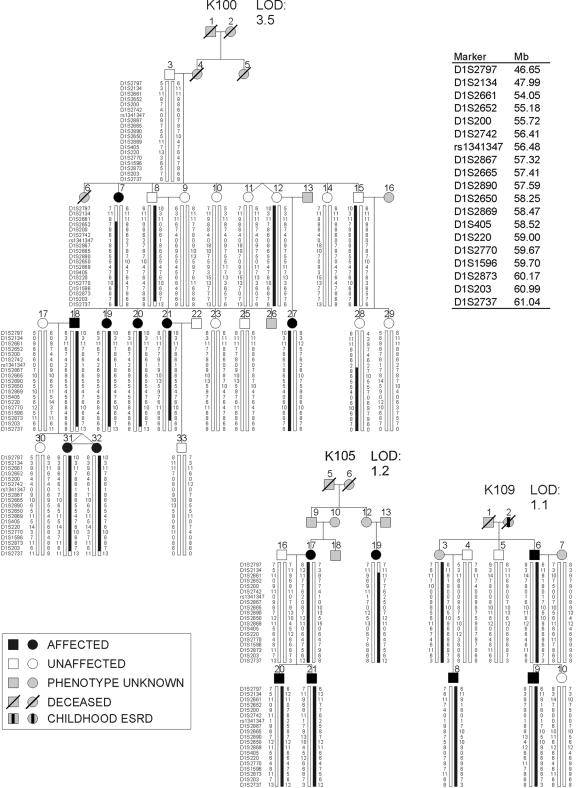
Nonsyndromic defects in the urinary tract are the most common cause of end-stage renal failure in children and account for a significant proportion of adult nephropathy. The genetic basis of these disorders is not fully understood. We studied seven multiplex kindreds ascertained via an index case with a nonsyndromic solitary kidney or renal hypodysplasia. Systematic ultrasonographic screening revealed that many family members harbor malformations, such as solitary kidneys, hypodysplasia, or ureteric abnormalities (in a total of 29 affected individuals). A genomewide scan identified significant linkage to a 6.9-Mb segment on chromosome 1p32-33 under an autosomal dominant model with reduced penetrance (peak LOD score 3.5 at D1S2652 in the largest kindred). Altogether, three of the seven families showed positive LOD scores at this interval, demonstrating heterogeneity of the trait (peak HLOD 3.9, with 45% of families linked). The chromosome 1p32-33 interval contains 52 transcription units, and at least 23 of these are expressed at stage E12.5 in the murine ureteric bud and/or metanephric mesenchyme. These data show that autosomal dominant nonsyndromic renal hypodysplasia and associated urinary tract malformations are genetically heterogeneous and identify a locus for this common cause of human kidney failure.
Figures
References
Web Resources
-
- Ensembl, http://www.ensembl.org/Homo_sapiens/index.html (for v39)
-
- NCBI Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/ (for build 36.1)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for renal agenesis/adysplasia, renal-coloboma syndrome, Fraser syndrome, and branchiootorenal syndrome)
-
- UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway (for March 2006 assembly)
References
-
- Helin I, Persson PH (1986) Prenatal diagnosis of urinary tract abnormalities by ultrasound. Pediatrics 78:879–883 - PubMed
-
- US Renal Data System (2005) USRDS 2005 annual data report: atlas of end-stage renal disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
