

Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship
- PMID: 17276014
- PMCID: PMC2288663
- DOI: 10.1016/j.neuroscience.2006.12.020
Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship
Abstract
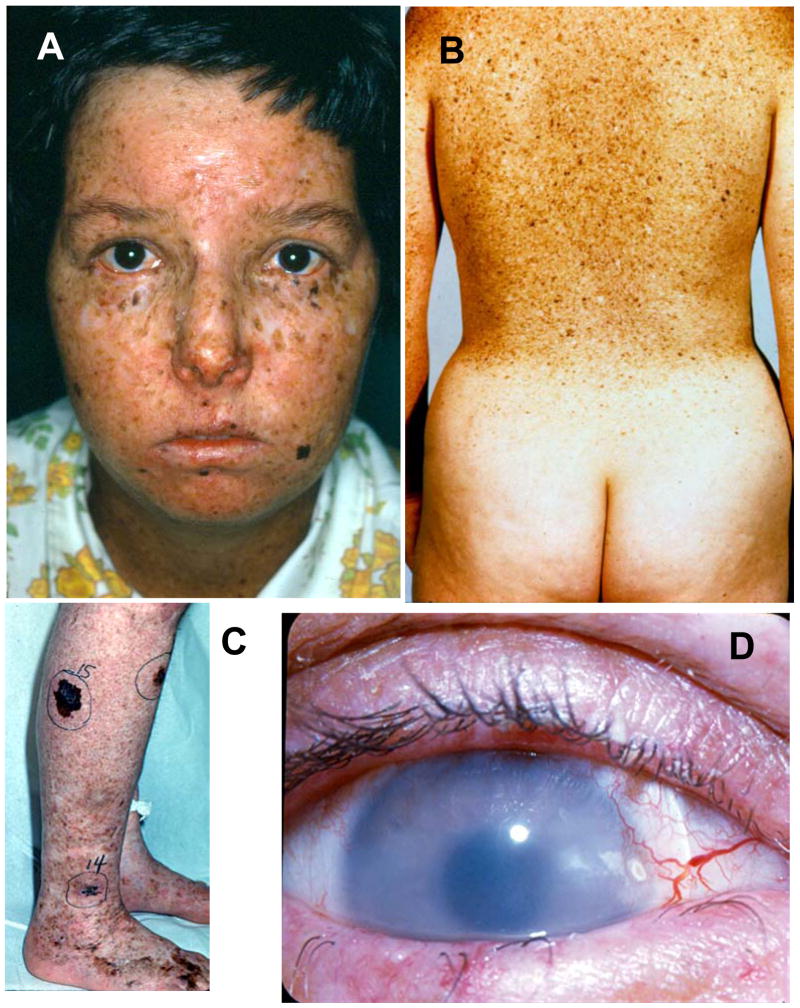
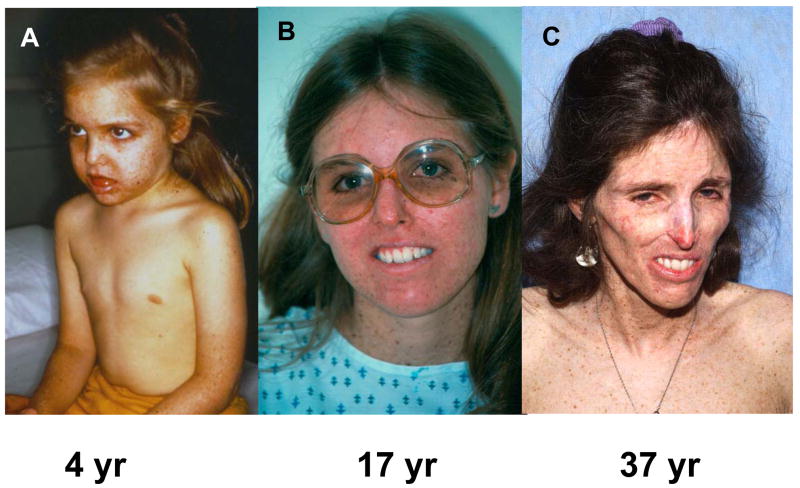
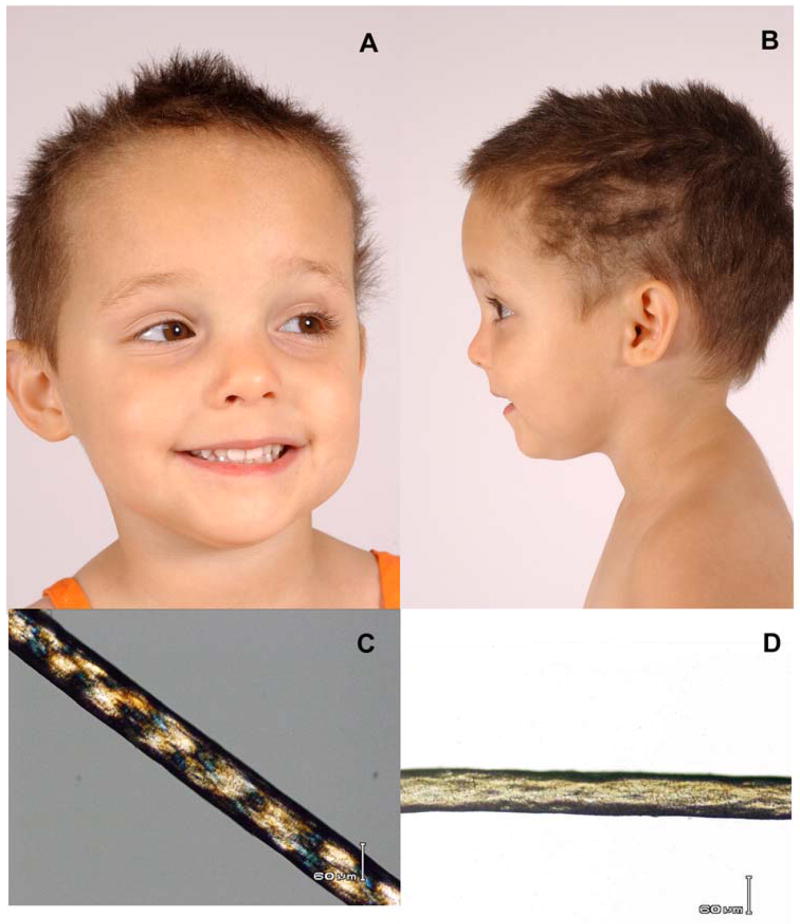
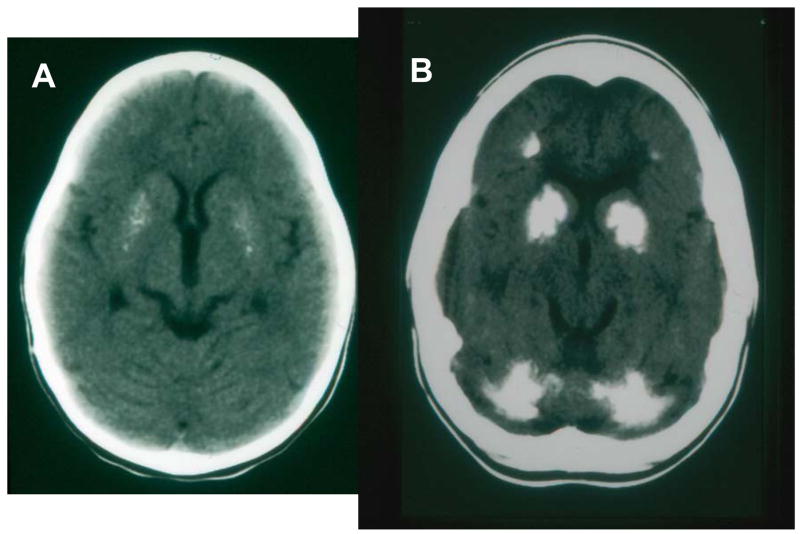
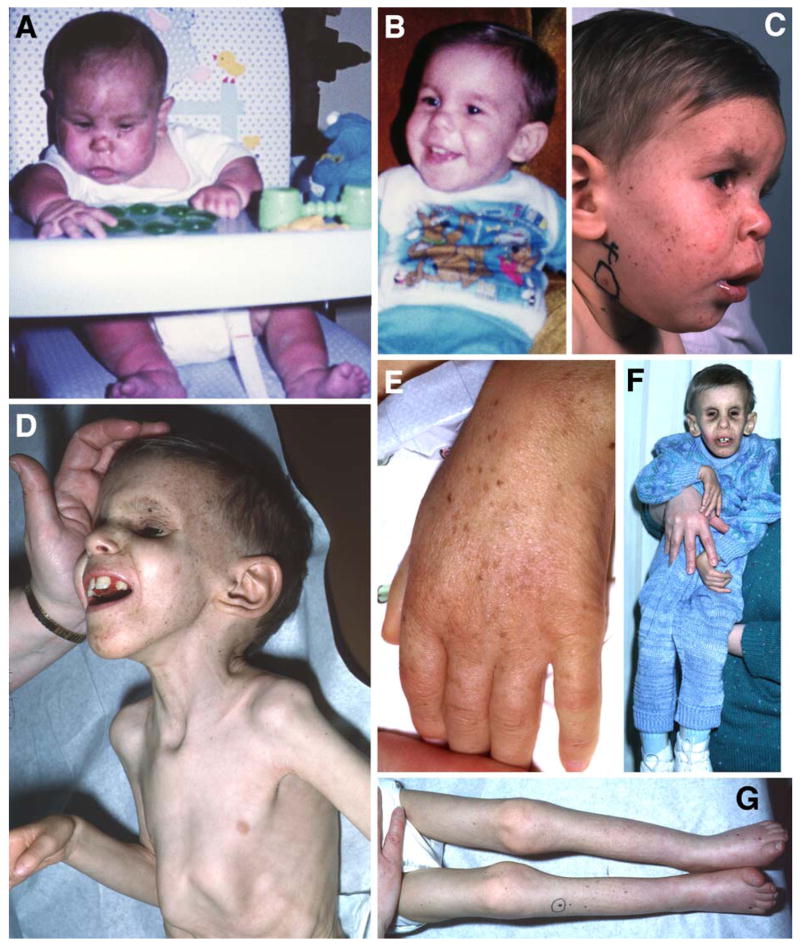
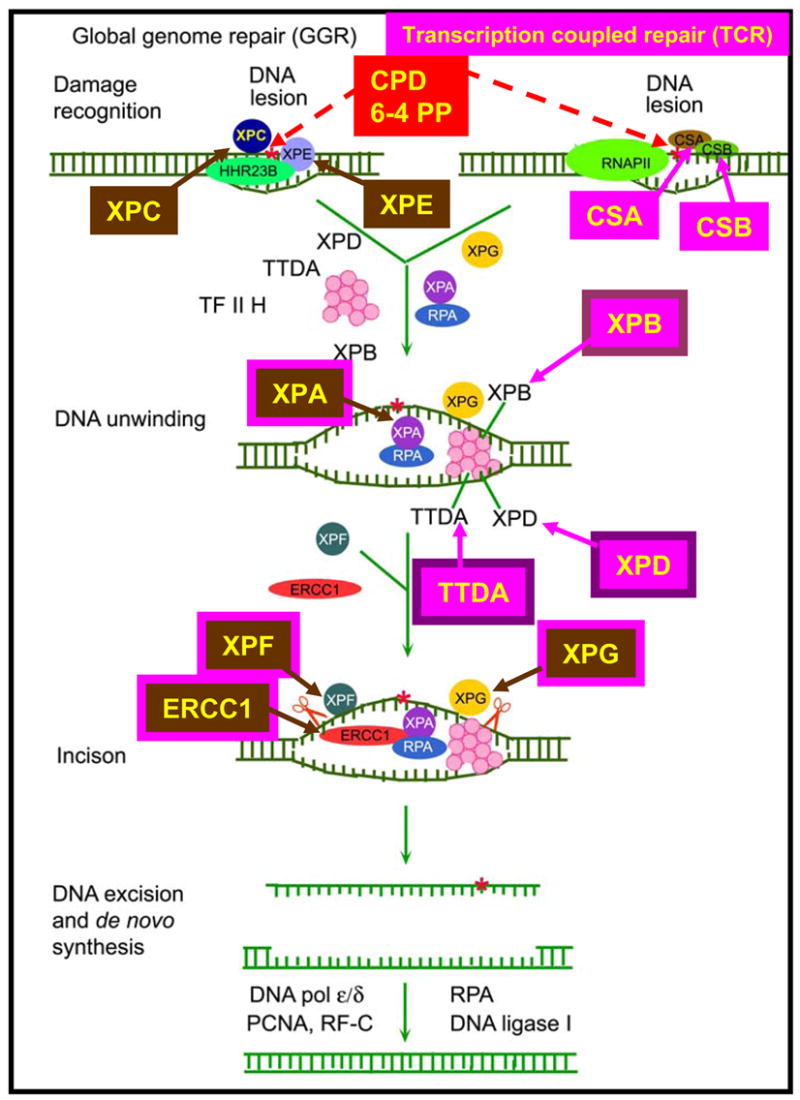
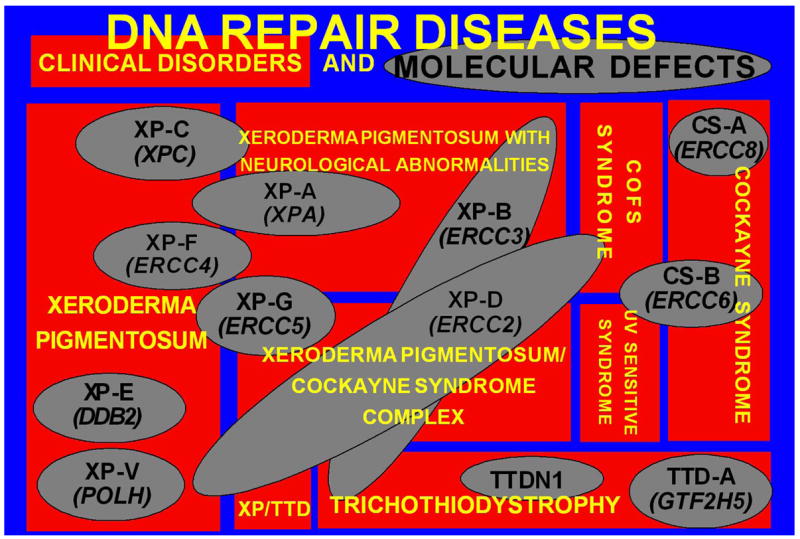
Patients with the rare genetic disorders, xeroderma pigmentosum (XP), trichothiodystrophy (TTD) and Cockayne syndrome (CS) have defects in DNA nucleotide excision repair (NER). The NER pathway involves at least 28 genes. Three NER genes are also part of the basal transcription factor, TFIIH. Mutations in 11 NER genes have been associated with clinical diseases with at least eight overlapping phenotypes. The clinical features of these patients have some similarities but also have marked differences. NER is involved in protection against sunlight-induced DNA damage. While XP patients have 1000-fold increase in susceptibility to skin cancer, TTD and CS patients have normal skin cancer risk. Several of the genes involved in NER also affect somatic growth and development. Some patients have short stature and immature sexual development. TTD patients have sulfur deficient brittle hair. Progressive sensorineural deafness is an early feature of XP and CS. Many of these clinical diseases are associated with developmental delay and progressive neurological degeneration. The main neuropathology of XP is a primary neuronal degeneration. In contrast, CS and TTD patients have reduced myelination of the brain. These complex neurological abnormalities are not related to sunlight exposure but may be caused by developmental defects as well as faulty repair of DNA damage to neuronal cells induced by oxidative metabolism or other endogenous processes.
Figures
References
-
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JHJ. Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. 2. McGraw-Hill; New York: 2002. pp. 211–237.
-
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. - PubMed
-
- Cleaver JE, Thompson LH, Richardson AS, States JC. A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat. 1999;14:9–22. - PubMed
-
- Dollfus H, Porto F, Caussade P, Speeg-Schatz C, Sahel J, Grosshans E, Flament J, Sarasin A. Ocular manifestations in the inherited DNA repair disorders. Surv Ophthalmol. 2003;48:107–122. - PubMed
-
- Dubaele S, Proietti d S, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
