

Identifying allosteric fluctuation transitions between different protein conformational states as applied to Cyclin Dependent Kinase 2
- PMID: 17286863
- PMCID: PMC1800904
- DOI: 10.1186/1471-2105-8-45
Identifying allosteric fluctuation transitions between different protein conformational states as applied to Cyclin Dependent Kinase 2
Abstract
Background: The mechanisms underlying protein function and associated conformational change are dominated by a series of local entropy fluctuations affecting the global structure yet are mediated by only a few key residues. Transitional Dynamic Analysis (TDA) is a new method to detect these changes in local protein flexibility between different conformations arising from, for example, ligand binding. Additionally, Positional Impact Vertex for Entropy Transfer (PIVET) uses TDA to identify important residue contact changes that have a large impact on global fluctuation. We demonstrate the utility of these methods for Cyclin-dependent kinase 2 (CDK2), a system with crystal structures of this protein in multiple functionally relevant conformations and experimental data revealing the importance of local fluctuation changes for protein function.
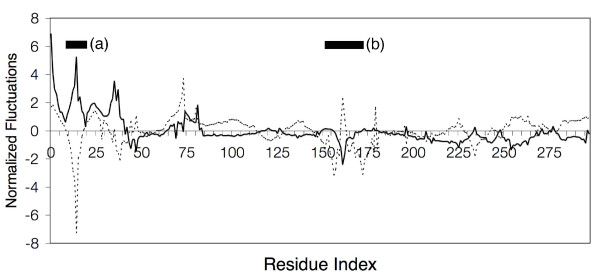
Results: TDA and PIVET successfully identified select residues that are responsible for conformation specific regional fluctuation in the activation cycle of Cyclin Dependent Kinase 2 (CDK2). The detected local changes in protein flexibility have been experimentally confirmed to be essential for the regulation and function of the kinase. The methodologies also highlighted possible errors in previous molecular dynamic simulations that need to be resolved in order to understand this key player in cell cycle regulation. Finally, the use of entropy compensation as a possible allosteric mechanism for protein function is reported for CDK2.
Conclusion: The methodologies embodied in TDA and PIVET provide a quick approach to identify local fluctuation change important for protein function and residue contacts that contributes to these changes. Further, these approaches can be used to check for possible errors in protein dynamic simulations and have the potential to facilitate a better understanding of the contribution of entropy to protein allostery and function.
Figures




Similar articles
-
Network-based modelling and percolation analysis of conformational dynamics and activation in the CDK2 and CDK4 proteins: dynamic and energetic polarization of the kinase lobes may determine divergence of the regulatory mechanisms.Mol Biosyst. 2017 Oct 24;13(11):2235-2253. doi: 10.1039/c7mb00355b. Mol Biosyst. 2017. PMID: 28926061
-
Insights on Structural Characteristics and Ligand Binding Mechanisms of CDK2.Int J Mol Sci. 2015 Apr 24;16(5):9314-40. doi: 10.3390/ijms16059314. Int J Mol Sci. 2015. PMID: 25918937 Free PMC article. Review.
-
Preferential binding of allosteric modulators to active and inactive conformational states of metabotropic glutamate receptors.BMC Bioinformatics. 2008;9 Suppl 1(Suppl 1):S16. doi: 10.1186/1471-2105-9-S1-S16. BMC Bioinformatics. 2008. PMID: 18315847 Free PMC article.
-
Probing the flexibility of large conformational changes in protein structures through local perturbations.PLoS Comput Biol. 2009 Apr;5(4):e1000343. doi: 10.1371/journal.pcbi.1000343. Epub 2009 Apr 3. PLoS Comput Biol. 2009. PMID: 19343225 Free PMC article.
-
Cyclin-Dependent Kinase 2 in Cellular Senescence and Cancer. A Structural and Functional Review.Curr Drug Targets. 2019;20(7):716-726. doi: 10.2174/1389450120666181204165344. Curr Drug Targets. 2019. PMID: 30516105 Review.
Cited by
-
Contact rearrangements form coupled networks from local motions in allosteric proteins.Proteins. 2008 Apr;71(1):455-66. doi: 10.1002/prot.21800. Proteins. 2008. PMID: 17957766 Free PMC article.
-
Discovery of a potential allosteric ligand binding site in CDK2.ACS Chem Biol. 2011 May 20;6(5):492-501. doi: 10.1021/cb100410m. Epub 2011 Feb 22. ACS Chem Biol. 2011. PMID: 21291269 Free PMC article.
-
Structure-based predictive models for allosteric hot spots.PLoS Comput Biol. 2009 Oct;5(10):e1000531. doi: 10.1371/journal.pcbi.1000531. Epub 2009 Oct 9. PLoS Comput Biol. 2009. PMID: 19816556 Free PMC article.
-
Loosening of Side-Chain Packing Associated with Perturbations in Peripheral Dynamics Induced by the D76N Mutation of β2-Microglobulin Revealed by Pressure-NMR and Molecular Dynamic Simulations.Biomolecules. 2019 Sep 16;9(9):491. doi: 10.3390/biom9090491. Biomolecules. 2019. PMID: 31527472 Free PMC article.
-
Dissecting conformational changes in APP's transmembrane domain linked to ε-efficiency in familial Alzheimer's disease.PLoS One. 2018 Jul 2;13(7):e0200077. doi: 10.1371/journal.pone.0200077. eCollection 2018. PLoS One. 2018. PMID: 29966005 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
