

FlowerPower: clustering proteins into domain architecture classes for phylogenomic inference of protein function
- PMID: 17288570
- PMCID: PMC1796606
- DOI: 10.1186/1471-2148-7-S1-S12
FlowerPower: clustering proteins into domain architecture classes for phylogenomic inference of protein function
Abstract
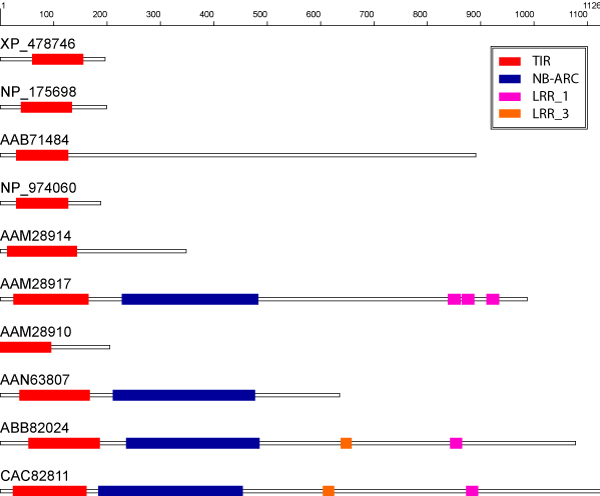
Background: Function prediction by transfer of annotation from the top database hit in a homology search has been shown to be prone to systematic error. Phylogenomic analysis reduces these errors by inferring protein function within the evolutionary context of the entire family. However, accuracy of function prediction for multi-domain proteins depends on all members having the same overall domain structure. By contrast, most common homolog detection methods are optimized for retrieving local homologs, and do not address this requirement.
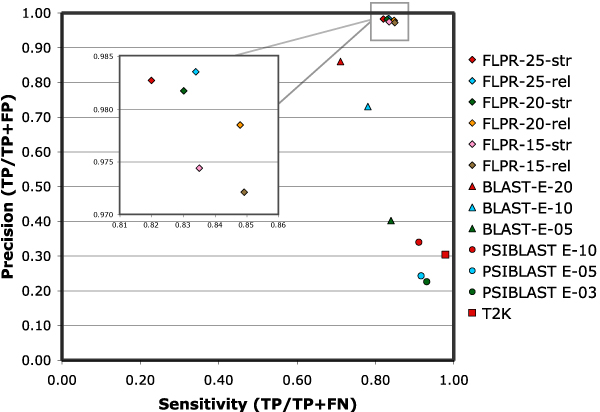
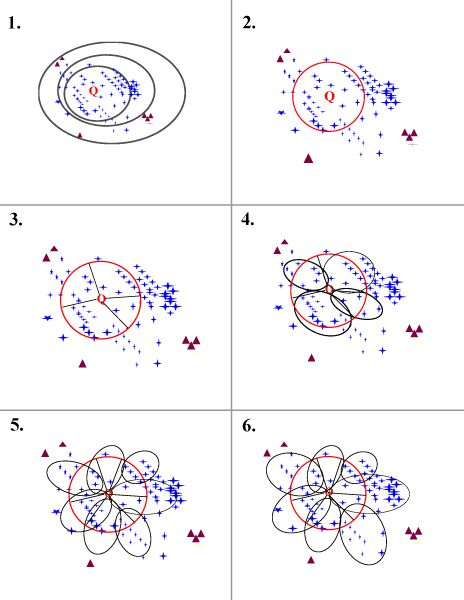
Results: We present FlowerPower, a novel clustering algorithm designed for the identification of global homologs as a precursor to structural phylogenomic analysis. Similar to methods such as PSIBLAST, FlowerPower employs an iterative approach to clustering sequences. However, rather than using a single HMM or profile to expand the cluster, FlowerPower identifies subfamilies using the SCI-PHY algorithm and then selects and aligns new homologs using subfamily hidden Markov models. FlowerPower is shown to outperform BLAST, PSI-BLAST and the UCSC SAM-Target 2K methods at discrimination between proteins in the same domain architecture class and those having different overall domain structures.
Conclusion: Structural phylogenomic analysis enables biologists to avoid the systematic errors associated with annotation transfer; clustering sequences based on sharing the same domain architecture is a critical first step in this process. FlowerPower is shown to consistently identify homologous sequences having the same domain architecture as the query.
Availability: FlowerPower is available as a webserver at http://phylogenomics.berkeley.edu/flowerpower/.
Figures


References
-
- Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–167. - PubMed
-
- Galperin MY, Koonin EV. Sources of systematic error in functional annotation of genomes: domain rearrangement, non-orthologous gene displacement and operon disruption. In Silico Biol. 1998;1:55–67. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
