

Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model
- PMID: 17288577
- PMCID: PMC1796613
- DOI: 10.1186/1471-2148-7-S1-S4
Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model
Abstract
Background: Thanks to the large amount of signal contained in genome-wide sequence alignments, phylogenomic analyses are converging towards highly supported trees. However, high statistical support does not imply that the tree is accurate. Systematic errors, such as the Long Branch Attraction (LBA) artefact, can be misleading, in particular when the taxon sampling is poor, or the outgroup is distant. In an otherwise consistent probabilistic framework, systematic errors in genome-wide analyses can be traced back to model mis-specification problems, which suggests that better models of sequence evolution should be devised, that would be more robust to tree reconstruction artefacts, even under the most challenging conditions.
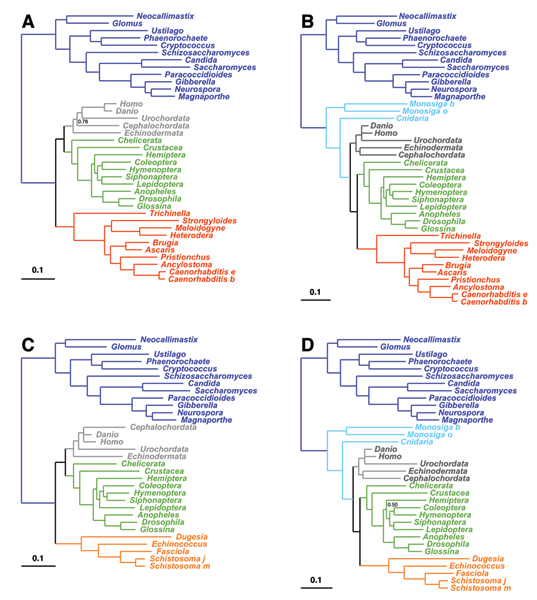
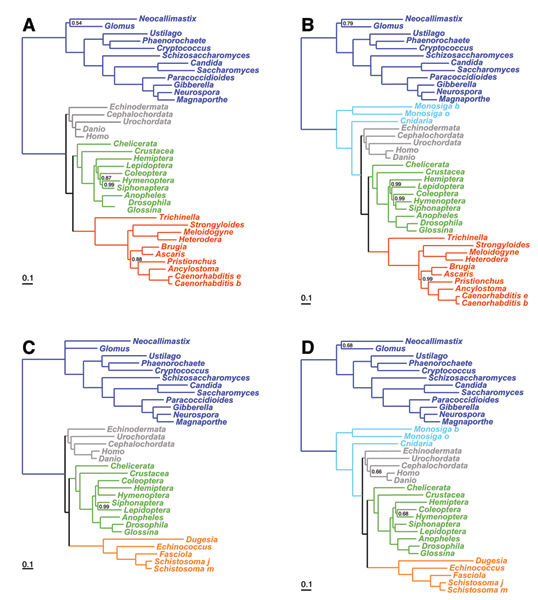
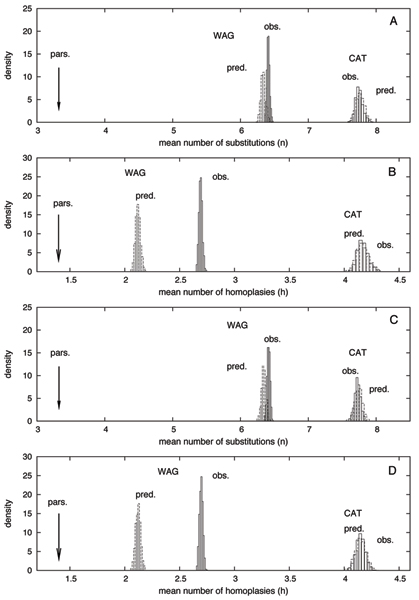
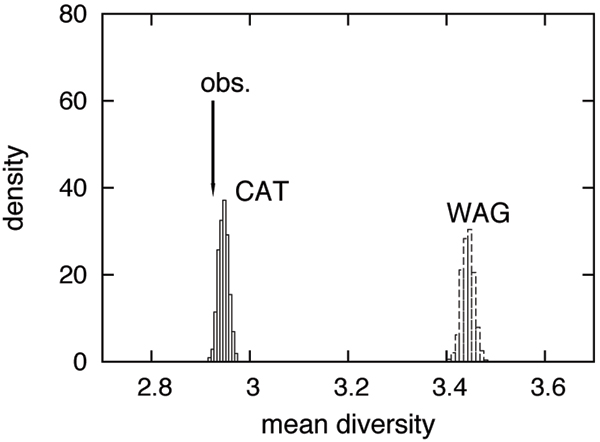
Methods: We focus on a well characterized LBA artefact analyzed in a previous phylogenomic study of the metazoan tree, in which two fast-evolving animal phyla, nematodes and platyhelminths, emerge either at the base of all other Bilateria, or within protostomes, depending on the outgroup. We use this artefactual result as a case study for comparing the robustness of two alternative models: a standard, site-homogeneous model, based on an empirical matrix of amino-acid replacement (WAG), and a site-heterogeneous mixture model (CAT). In parallel, we propose a posterior predictive test, allowing one to measure how well a model acknowledges sequence saturation.
Results: Adopting a Bayesian framework, we show that the LBA artefact observed under WAG disappears when the site-heterogeneous model CAT is used. Using cross-validation, we further demonstrate that CAT has a better statistical fit than WAG on this data set. Finally, using our statistical goodness-of-fit test, we show that CAT, but not WAG, correctly accounts for the overall level of saturation, and that this is due to a better estimation of site-specific amino-acid preferences.
Conclusion: The CAT model appears to be more robust than WAG against LBA artefacts, essentially because it correctly anticipates the high probability of convergences and reversions implied by the small effective size of the amino-acid alphabet at each site of the alignment. More generally, our results provide strong evidence that site-specificities in the substitution process need be accounted for in order to obtain more reliable phylogenetic trees.
Figures
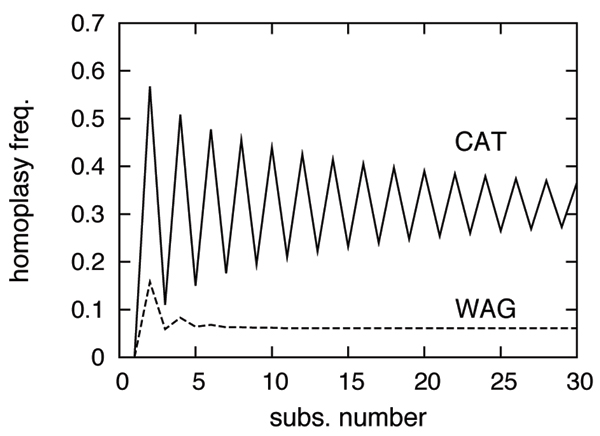
References
-
- Philippe H, Delsuc F, Brinkmann H, Lartillot N. Phylogenomics. Annu Rev Ecol Evol Syst. 2005;36:541–562. doi: 10.1146/annurev.ecolsys.35.112202.130205. - DOI
-
- Kluge AG. A concern for evidence and a phylogenetic hypothesis if relationships among Epicrates (Boidae, Serpentes) Syst Zool. 1989;38:7–25. doi: 10.2307/2992432. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
