

A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway
- PMID: 17289571
- PMCID: PMC1847584
- DOI: 10.1016/j.cell.2006.12.032
A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway
Abstract
Yeast cells arrest in the G1 phase of the cell cycle upon exposure to mating pheromones. As cells commit to a new cycle, G1 CDK activity (Cln/CDK) inhibits signaling through the mating MAPK cascade. Here we show that the target of this inhibition is Ste5, the MAPK cascade scaffold protein. Cln/CDK disrupts Ste5 membrane localization by phosphorylating a cluster of sites that flank a small, basic, membrane-binding motif in Ste5. Effective inhibition of Ste5 signaling requires multiple phosphorylation sites and a substantial accumulation of negative charge, which suggests that Ste5 acts as a sensor for high G1 CDK activity. Thus, Ste5 is an integration point for both external and internal signals. When Ste5 cannot be phosphorylated, pheromone triggers an aberrant arrest of cells outside G1 either in the presence or absence of the CDK-inhibitor protein Far1. These findings define a mechanism and physiological benefit of restricting antiproliferative signaling to G1.
Figures

Pheromone response pathway, showing membrane recruitment of Ste5.
Methods for activating membrane-localized signaling. From left to right: α factor (αf) treatment or Gβ overexpression (Whiteway et al., 1990); hyperactive membrane localization of Ste5 via an enhanced PM domain (Winters et al., 2005); membrane targeting of Ste5 via a foreign transmembrane domain (Pryciak and Huntress, 1998); membrane targeting of Ste11 via a prenylation/palmitoylation motif (Winters et al., 2005).
Cln2/CDK inhibition correlates with dependence on the Ste5 PM domain. Pathway-activating components were expressed from the GAL1 promoter and compared for their ability to induce FUS1-lacZ transcription in ste4Δ strains ± PGAL1-CLN2 (n = 4).
Ste20-independent signaling is sensitive to Cln2 inhibition. Wild-type Ste11 (WT) or a Ste20-independent mutant (Ste11-Asp3) was expressed in ste11Δ or ste11Δ ste20Δ strains ± PGAL1-CLN2. FUS1-lacZ induction was measured after α factor treatment (n = 6).
PM domain mutations that disrupt nuclear targeting (NLSm) do not affect Cln2 inhibition. FUS1-lacZ was induced by α factor treatment in ste5Δ ± PGAL1-CLN2 strains expressing Ste5-WT or Ste5-NLSm (n = 9).
Increased Ste5 membrane affinity causes increased resistance to Cln2. Left, Ste5 variants contained PM domain mutations that increase membrane affinity. Right, the native PM domain was replaced with 1 or 2 copies of the PLCδ PH domain. All forms were expressed from the native STE5 promoter in ste5Δ strains ± PGAL1-CLN2, and response to α factor was measured (n = 4-9).
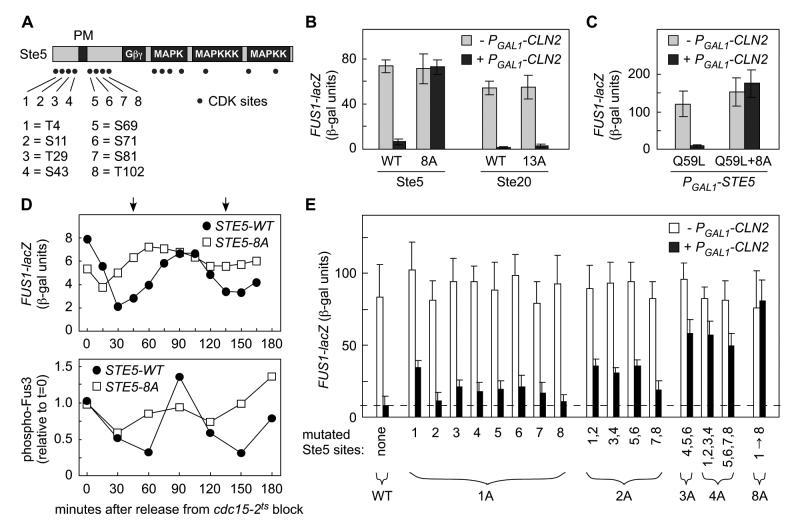
Locations of potential CDK phosphorylation sites (SP or TP) in Ste5.
Elimination of 8 N-terminal CDK sites in Ste5 (Ste5-8A) causes resistance to Cln2.
Response to α factor was measured in ste5Δ ± PGAL1-CLN2 cells expressing Ste5 variants (WT or 8A) from the native STE5 promoter, or in ste20Δ ± PGAL1-CLN2 cells expressing Ste20 variants (WT or 13A) from the native STE20 promoter. Bars, mean ± SD (n = 8).
CDK resistance caused by 8A mutations restores membrane signaling independent of Ste5-Gβγ interaction. Gβγ-independent signaling was activated by PGAL1-STE5-Q59L ± 8A in ste4Δ ste5Δ cells ± PGAL1-CLN2. Bars, mean ± SD (n = 7).
The Ste5-8A mutant disrupts cell cycle periodicity of pheromone response. Cells (cdc15-2 or cdc15-2 STE5-8A) were synchronized in late M phase by arrest at 36°C, and then transferred to 25°C. At various times, response to brief treatment with α factor was monitored (see Experimental Procedures). Top, FUS1-lacZ induction (mean of 4 trials). Bottom, Fus3 activation (phospho-Fus3) was measured using phospho-specific antibodies (mean of 6 trials). Arrows mark the times of bud emergence (see Figures S1D and 5G).
Ste5 phosphorylation sites were replaced with Ala residues either singly (1A) or in various combinations (2A, 3A, 4A, 8A). Response to α factor was tested in ste5Δ strains ± PGAL1-CLN2 (mean + SD, n = 8-16).

Glu replacement mutations at CDK sites. S/T residues were replaced with E or A, and SP/TP dipeptides were replaced with EE or AA, as indicated.
Inhibition of Ste5 signaling is proportional to added negative charge. Ste5 mutants were tested for α factor response in ste5Δ cells ± PGAL1-CLN2. Ste5 “up4A” and “dn4A” refer to Ala mutations at sites #1-4 and #5-8, respectively. Bars, FUS1-lacZ levels, relative to Ste5-WT (mean ± SD, n = 6).
Anti-myc blot showing levels of Ste5-myc13 mutants expressed in ste5Δ cells.
The Ste5-16E mutant can still bind Ste4. Extracts of ste4Δ ste5Δ cells coexpressing Ste5-myc and GFP-Ste4 (after 3 hr induction of PGAL1-GFP-STE4) were analyzed by immunoprecipitation (IP) and immunoblotting (blot) as indicated. Ste5-C180A served as a control that is defective at binding Ste4 (Feng et al., 1998).
Ste5 Glu mutants are competent to mediate basal signaling (i.e., no α factor) activated by Ste11-4 in ste4Δ ste5Δ ste20Δ cells. Bars, mean ± SD (n = 4).
The 16E mutations only inhibit signaling that requires the Ste5 PM domain. FUS1-lacZ (mean ± SD, n = 3-6) was induced in ste4Δ ste5Δ cells (without α factor) by PGAL1-driven expression of Ste5, Ste5-Q59L, or Ste5-CTM, each of which either contained the 16E mutations (+16E) or did not (−16E). Anti-GFP blots confirmed that protein levels were unaffected by the 16E mutations (data not shown).
Glu mutants disrupt Gβγ-independent, membrane-localized signaling. Signaling (mean ± SD, n = 6) was activated in ste4Δ ste5Δ cells (without α factor) by coexpression of PGAL1-STE11-Cpr with the indicated Ste5 derivatives.
Ste5 derivatives containing various Ala or Glu mutations were coexpressed in ste4Δ ste5Δ cells with either Ste4-WT or Ste4 mutants (K55E or N157H S175P) that weaken Ste5 binding (Leeuw et al., 1998; Winters et al., 2005). Response to α factor was measured (mean ± SD, n=6).

Localization of GFP-Ste5-Q59L ± 8A, expressed from the GAL1 promoter in ste4Δ ste7Δ cells ± PGAL1-CLN2. Note that hyperpolarized bud growth is due to Cln2 overexpression, not mating signaling. Also see Figure S2B.
The 16E mutations disrupt Ste5 membrane localization mediated by the PM domain (Q59L), but not that mediated by a foreign transmembrane domain (CTM). Top, membrane localization induces mating pathway signaling, causing pear-shaped “shmoo” morphology. Bottom, localization results were similar in a non-signaling strain (ste4Δ ste7Δ).
Negative charge disrupts membrane localization of Ste5 N-terminal fragments. Localization was compared (in ste4Δ ste7Δ cells) for WT and mutant derivatives of GFP-Ste5(1-214) and GST-GFP-Ste5(1-125), which can localize to the membrane in the absence of pheromone, Gβγ, and other Ste5 sequences (Winters et al., 2005).

Phosphorylation of the Ste5 N-terminus by Cln2/Cdc28 in vitro. Bacterially-expressed GST-Ste51-125 fusions (WT, 8A, and 8E) were phosphorylated by recombinant Cln2-Cdc28. Histone H1 served as a control substrate.
Cln2 expression in vivo alters Ste5 electrophoretic mobility. HA-tagged Ste5 (WT, 8A, and 16E) was immunoprecipitated from the indicated strains after 3 hr galactose induction (to express Cln2), resolved by SDS-PAGE, and analyzed by anti-HA immunoblot.
The Ste5 mobility shift is due to phosphorylation. Ste5-HA3 was immunoprecipitated from ste5Δ ± PGAL1-CLN2 strains, and treated with calf intestinal phosphatase (CIP).
Effects on Ste5 mobility are specific to Cln2. Ste5-HA3 derivatives (WT, 8A, ΔNLS, ΔNLS+8A) were analyzed as in panel B, using various PGAL1-cyclin strains.
Ste5ΔNLS is more fully modified by Cln2. Ste5-HA3 derivatives were analyzed as in panel B.
Ste5ΔNLS modification is elevated after Start and requires Cln1/2. WT and mutant strains (“cln1,2” = cln1 cln2) expressing Ste5-HA3 (ΔNLS or ΔNLS+8A) were incubated for 3 hr at 37°C.
Modification of the Ste5 N-terminus is cell cycle dependent. Cells (cdc15-2 or cdc28-13) harboring Ste5-HA3 (ΔNLS or ΔNLS+8A) were arrested at 37°C for 3 hr, then transferred to 25°C to resume cycling. Samples were collected at 20 min intervals (0-180 min). As cdc15 cells arrest with large buds, emergence of small buds was used to follow cell cycle progression (c.f., Figure S1D).

Pheromone arrests some STE5-8A cells at a post-Start (2N) stage. Cells were treated with α factor for the indicated times. FACS profiles show DNA content.
The post-Start arrest phenotype is dominant and reflects the level of CDK-resistance of Ste5. Wild type cells harboring STE5 plasmids (1A#5 = site #5; 1A#1 = site #1; 2A = sites #5-6) were treated with α factor for 3.25 hrs. The percent of cells with 2N DNA content was quantified by FACS (mean ± SD; n = 4). The dashed line marks the %2N value observed when the STE5-WT plasmid is present in wild-type cells.
G1 phase STE5-8A cells were purified by centrifugal elutriation, and treated with α factor either immediately or after cells resumed cycling. Arrest phenotypes were then compared. See Figures S3 and S4 for the complete data set. Arrowheads, cell buds with α factor-induced projections.
STE5-8A allows near-uniform 2N arrest when G1 arrest is bypassed using PGPD1-CLB5. Left, DNA content of cells after 3 hr ± α factor. Right, halo assays showing growth arrest by α factor.
Halo assays showing that STE5-8A restores pheromone arrest to far1Δ cells.
Suppression of the far1Δ arrest defect increases as more CDK sites are eliminated from Ste5. Fivefold serial dilutions of strains harboring STE5 plasmids (1A = site #5; 2A = sites #5-6; 4A = sites #1-4) were incubated on –Ura plates ± 1 μM α factor.
Pathway activation by CDK-resistant constructs causes Far1-independent arrest. Deletion strains harboring PGAL1-regulated activators of the mating pathway were grown on –Ura glucose or galactose plates.
The post-Start arrest triggered by Ste5-8A signaling is independent of Far1. The indicated strains were analyzed in parallel with those in panel D.

As cells pass Start, G1 CDK activity inhibits pheromone signaling by phosphorylating CDK sites flanking the PM domain in Ste5. The negatively-charged phosphates interfere with binding between the basic PM domain and the anionic phospholipid membrane.
Ste5 serves as an integration point for both external and internal regulatory cues, which act through the Gβγ-binding domain and the membrane-binding domain.
Far1 promotes pheromone arrest by inhibiting Cln/CDK activity, but Far1-independent arrest pathways also exist and are revealed when Ste5 signaling cannot be downregulated by Cln/CDK activity.
Comment in
-
Tuning bulk electrostatics to regulate protein function.Cell. 2007 Feb 9;128(3):441-4. doi: 10.1016/j.cell.2007.01.018. Cell. 2007. PMID: 17289565
References
-
- Bhattacharyya RP, Remenyi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. - PubMed
-
- Breitkreutz A, Boucher L, Tyers M. MAPK specificity in the yeast pheromone response independent of transcriptional activation. Curr Biol. 2001;11:1266–1271. - PubMed
-
- Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. Cell. 1990;63:999–1011. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
