

Subcellular localization of Grb2 by the adaptor protein Dok-3 restricts the intensity of Ca2+ signaling in B cells
- PMID: 17290227
- PMCID: PMC1852829
- DOI: 10.1038/sj.emboj.7601557
Subcellular localization of Grb2 by the adaptor protein Dok-3 restricts the intensity of Ca2+ signaling in B cells
Abstract
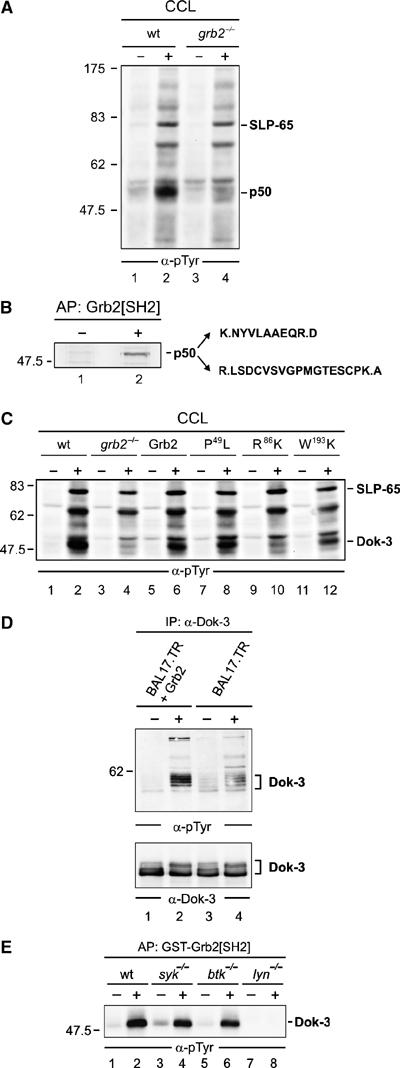
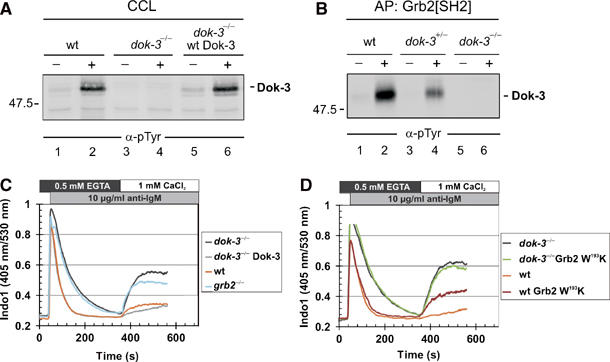
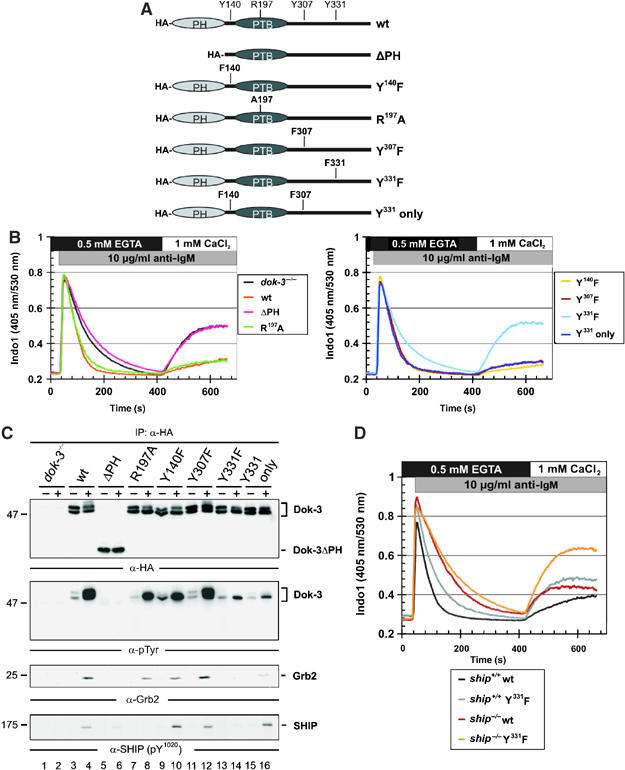
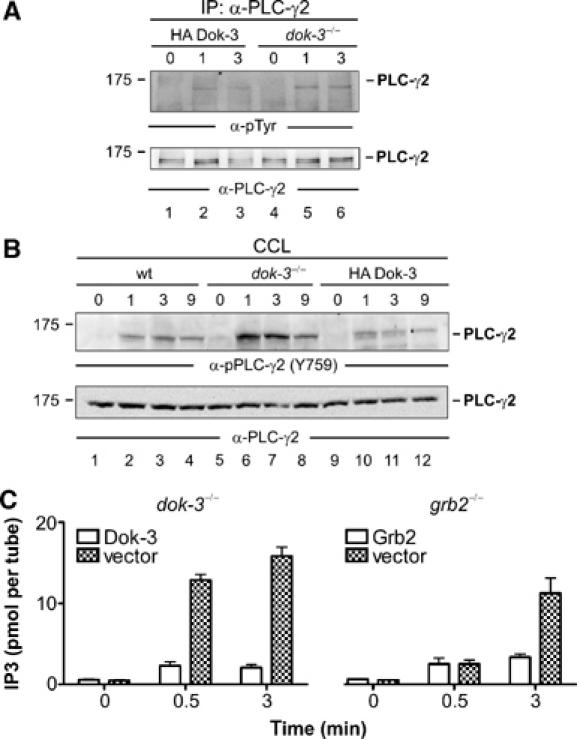
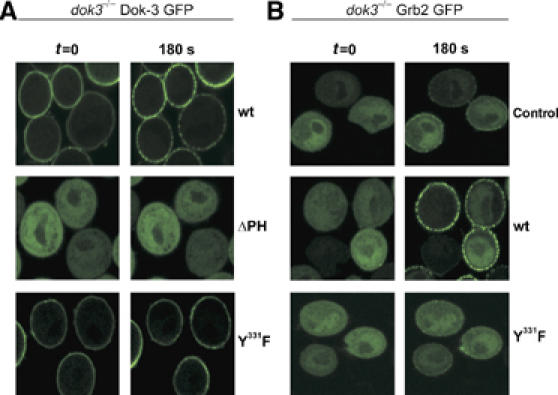
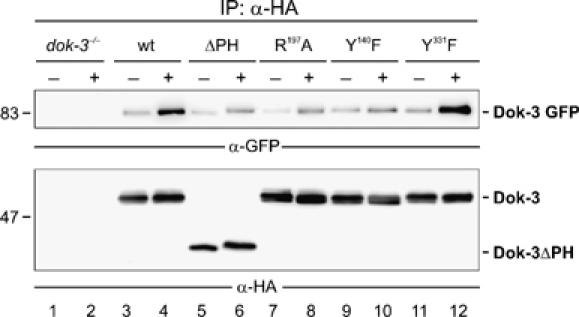
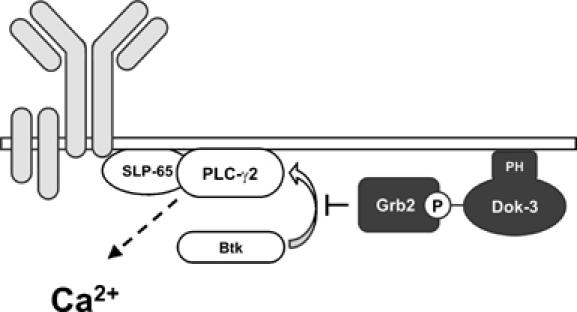
Spatial and temporal modulation of intracellular Ca2+ fluxes controls the cellular response of B lymphocytes to antigen stimulation. Herein, we identify the hematopoietic adaptor protein Dok-3 (downstream of kinase-3) as a key component of negative feedback regulation in Ca2+ signaling from the B-cell antigen receptor. Dok-3 localizes at the inner leaflet of the plasma membrane and is a major substrate for activated Src family kinase Lyn. Phosphorylated Dok-3 inhibits antigen receptor-induced Ca2+ elevation by recruiting cytosolic Grb2, which acts at this location as a negative regulator of Bruton's tyrosine kinase. This leads to diminished activation of phospholipase C-gamma2 and reduced production of soluble inositol trisphosphate. Hence, the Dok-3/Grb2 module is a membrane-associated signaling organizer, which orchestrates the interaction efficiency of Ca2+-mobilizing enzymes.
Figures
References
-
- Adachi T, Wienands J, Wakabayashi C, Yakura H, Reth M, Tsubata T (2001) SHP-1 requires inhibitory co-receptors to down-modulate B cell antigen receptor-mediated phosphorylation of cellular substrates. J Biol Chem 276: 26648–26655 - PubMed
-
- Bolland S, Pearse RN, Kurosaki T, Ravetch JV (1998) SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity 8: 509–516 - PubMed
-
- Boulay I, Nemorin JG, Duplay P (2005) Phosphotyrosine binding-mediated oligomerization of downstream of tyrosine kinase (Dok)-1 and Dok-2 is involved in CD2-induced Dok phosphorylation. J Immunol 175: 4483–4489 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
