

Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability
- PMID: 17304220
- PMCID: PMC1817623
- DOI: 10.1038/sj.emboj.7601574
Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability
Abstract
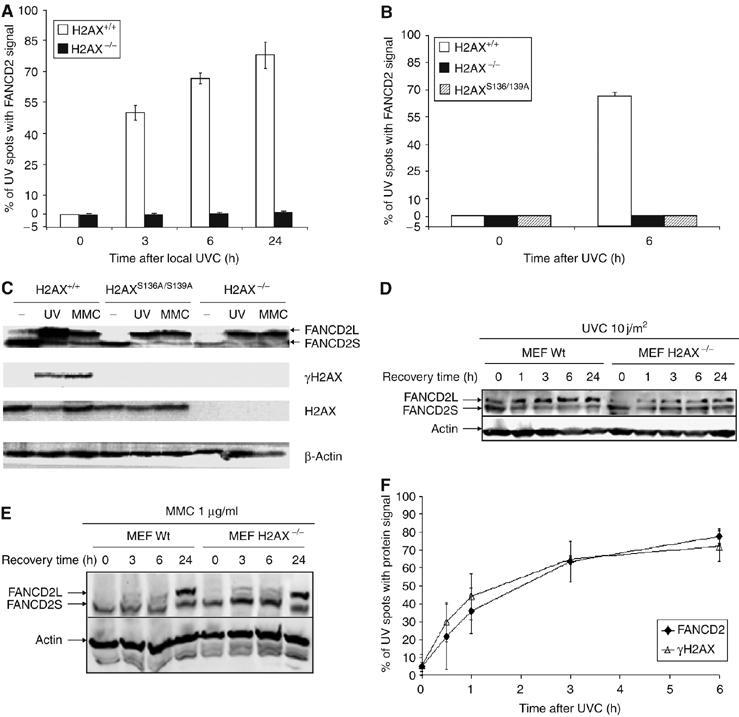
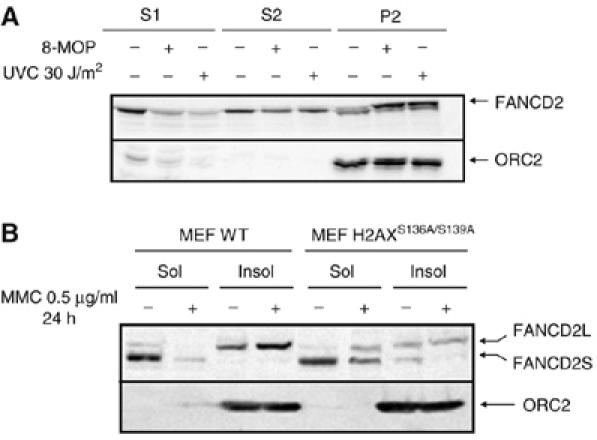
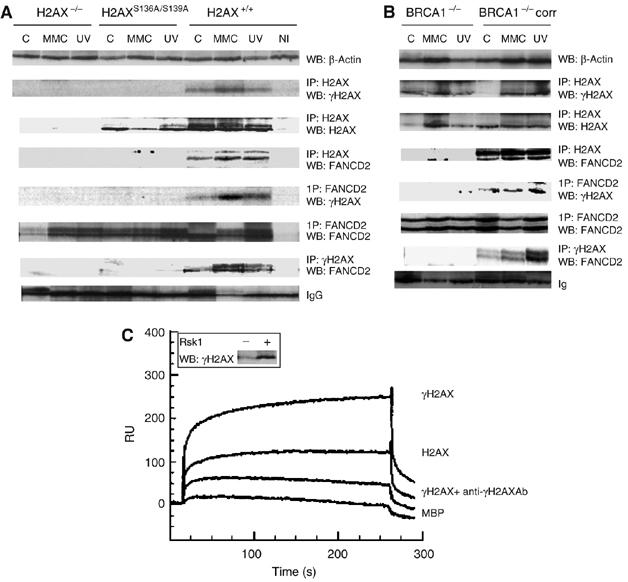
Fanconi anemia (FA) is a chromosome fragility syndrome characterized by bone marrow failure and cancer susceptibility. The central FA protein FANCD2 is known to relocate to chromatin upon DNA damage in a poorly understood process. Here, we have induced subnuclear accumulation of DNA damage to prove that histone H2AX is a novel component of the FA/BRCA pathway in response to stalled replication forks. Analyses of cells from H2AX knockout mice or expressing a nonphosphorylable H2AX (H2AX(S136A/S139A)) indicate that phosphorylated H2AX (gammaH2AX) is required for recruiting FANCD2 to chromatin at stalled replication forks. FANCD2 binding to gammaH2AX is BRCA1-dependent and cells deficient or depleted of H2AX show an FA-like phenotype, including an excess of chromatid-type chromosomal aberrations and hypersensitivity to MMC. This MMC hypersensitivity of H2AX-deficient cells is not further increased by depleting FANCD2, indicating that H2AX and FANCD2 function in the same pathway in response to DNA damage-induced replication blockage. Consequently, histone H2AX is functionally connected to the FA/BRCA pathway to resolve stalled replication forks and prevent chromosome instability.
Figures



References
-
- Abraham RT (2001) Cell cycle checkpoint signalling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 - PubMed
-
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114: 359–370 - PubMed
-
- Bassing CH, Alt FW (2004) H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 3: 149–153 - PubMed
-
- Bogliolo M, Taylor RM, Caldecott KW, Frosina G (2000) Reduced ligation during DNA base excision repair supported by BRCA2 mutant cells. Oncogene 19: 5781–5787 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
