

Case Reports
doi: 10.1136/jnnp.2006.104372.
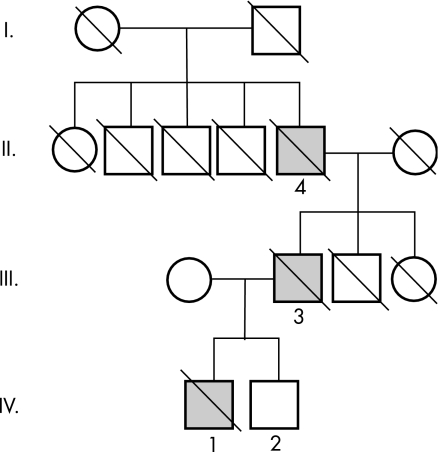
Familial prion disease in a Hungarian family with a novel 144-base pair insertion in the prion protein gene
Affiliations
- PMID: 17308293
- PMCID: PMC2117636
- DOI: 10.1136/jnnp.2006.104372
Item in Clipboard
Case Reports
Familial prion disease in a Hungarian family with a novel 144-base pair insertion in the prion protein gene
J Neurol Neurosurg Psychiatry.
2007 Mar.
Abstract
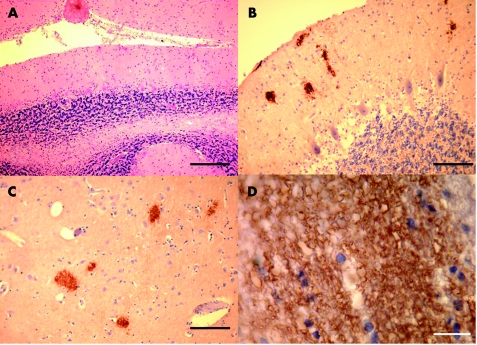
About 15% of human prion diseases are inherited, and are associated with point or insertional mutations of the prion protein gene (PRNP). Four families with six octapeptide repeat insertions (OPRI) in the PRNP gene have been described in the literature so far. Here we report two cases in a Hungarian family with a new six OPRI (R1R2R2R3R2R3gR3R2R2R3R4) in the PRNP gene. The clinical features (progressive ataxia, dementia and anosmia), the age of onset and the duration of disease were almost identical. In addition to the cerebellar and parahippocampal pathological changes already described, we also found deposits of pathological prion protein in the olfactory system.
Conflict of interest statement
Competing interests: None declared.
References
-
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 200124519–550. - PubMed
-
- King A, Doey L, Rossor M.et al Phenotypic variability in the brains of a family with a prion disease chracterized by a 144‐base pair insertion in the prion protein gene. Neuropathol Appl Neurobiol 20032998–105. - PubMed
-
- Vital C, Gray F, Vital A.et al Prion encephalopathy with insertion of octapeptide repeats: the number of repeats determines the type of cerebellar deposits. Neuropathol Appl Neurobiol 199824125–130. - PubMed
-
- Beck J A, Mead S, Campbell T A.et al Two‐octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology 200157354–356. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources