

Glycogen storage disease types I and II: treatment updates
- PMID: 17308886
- PMCID: PMC2692363
- DOI: 10.1007/s10545-007-0519-9
Glycogen storage disease types I and II: treatment updates
Abstract
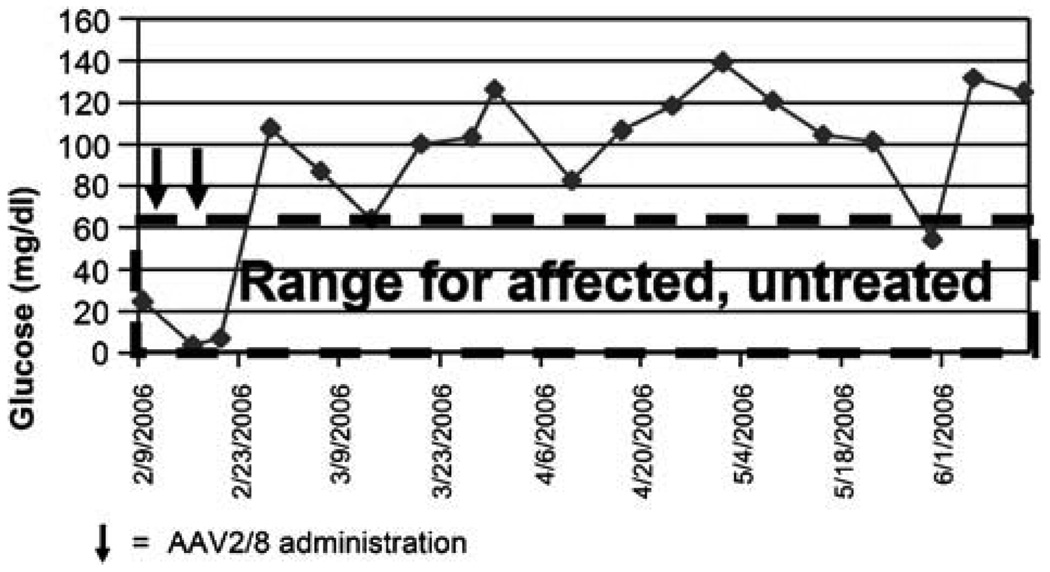
Prior to 2006 therapy for glycogen storage diseases consisted primarily of dietary interventions, which in the case of glycogen storage disease (GSD) type II (GSD II; Pompe disease) remained essentially palliative. Despite improved survival and growth, long-term complications of GSD type I (GSD I) have not responded to dietary therapy with uncooked cornstarch or continuous gastric feeding. The recognized significant risk of renal disease and liver malignancy in GSD I has prompted efforts towards curative therapy, including organ transplantation, in those deemed at risk. Results of clinical trials in infantile Pompe disease with alglucosidase alfa (Myozyme) showed prolonged survival reversal of cardiomyopathy, and motor gains. This resulted in broad label approval of Myozyme for Pompe disease in 2006. Furthermore, the development of experimental therapies, such as adeno-associated virus (AAV) vector-mediated gene therapy, holds promise for the availability of curative therapy in GSD I and GSD II/Pompe disease in the future.
Figures
References
-
- Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. - PubMed
-
- An Y, Young SP, Kishnani PS, et al. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab. 2005;85:247–254. - PubMed
-
- Beaty RM, Jackson M, Peterson D, et al. Delivery of glucose-6-phosphatase in a canine model for glycogen storage disease, type Ia, with adeno-associated virus (AAV) vectors. Gene Ther. 2002;9:1015–1022. - PubMed
-
- Bijvoet AG, Van Hirtum H, Kroos MA, et al. Human acid alphaglucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Hum Mol Genet. 1999;8:2145–2153. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
