

Blast sampling for structural and functional analyses
- PMID: 17319945
- PMCID: PMC1819393
- DOI: 10.1186/1471-2105-8-62
Blast sampling for structural and functional analyses
Abstract
Background: The post-genomic era is characterised by a torrent of biological information flooding the public databases. As a direct consequence, similarity searches starting with a single query sequence frequently lead to the identification of hundreds, or even thousands of potential homologues. The huge volume of data renders the subsequent structural, functional and evolutionary analyses very difficult. It is therefore essential to develop new strategies for efficient sampling of this large sequence space, in order to reduce the number of sequences to be processed. At the same time, it is important to retain the most pertinent sequences for structural and functional studies.
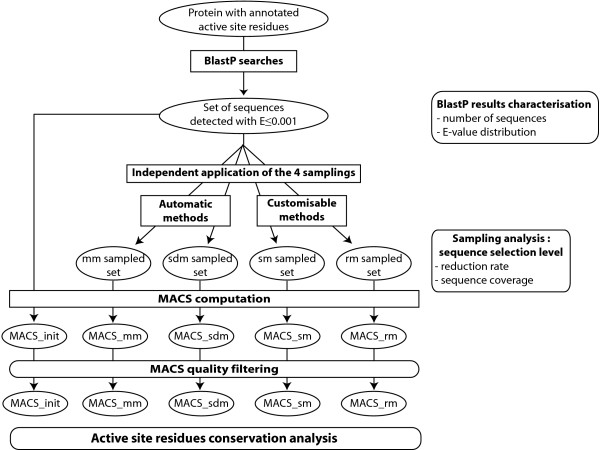
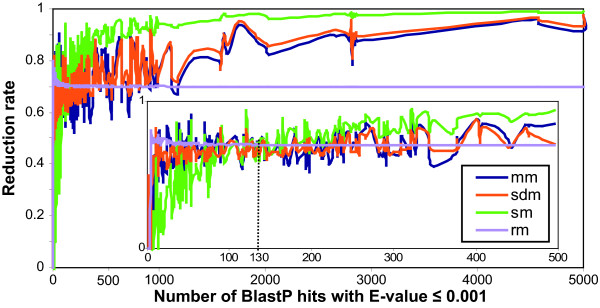
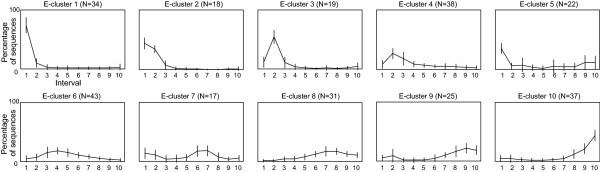
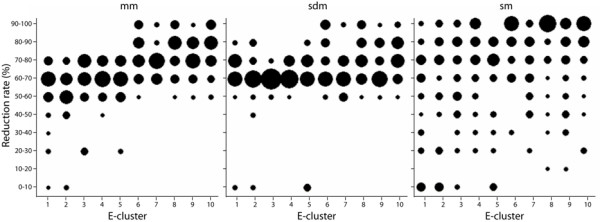
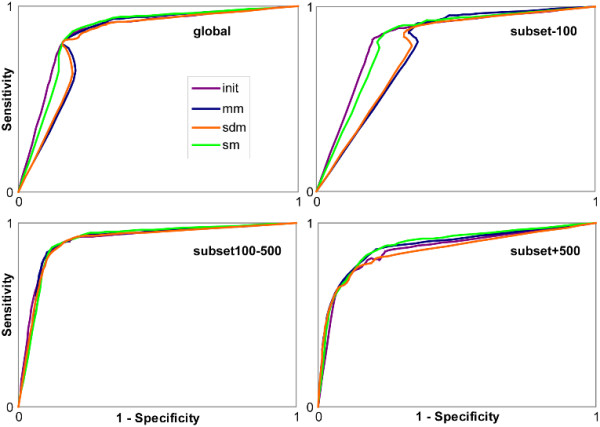
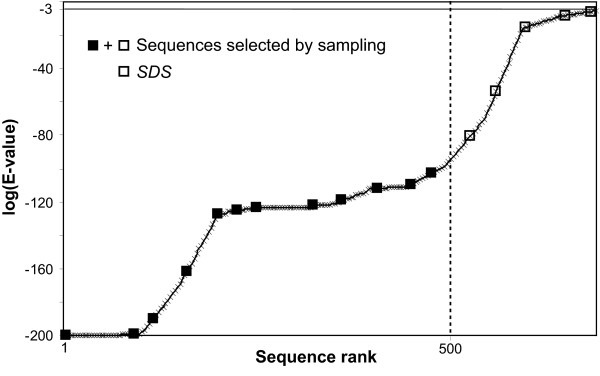
Results: An exhaustive analysis on a large scale test set (284 protein families) was performed to compare the efficiency of four different sampling methods aimed at selecting the most pertinent sequences. These four methods sample the proteins detected by BlastP searches and can be divided into two categories: two customisable methods where the user defines either the maximal number or the percentage of sequences to be selected; two automatic methods in which the number of sequences selected is determined by the program. We focused our analysis on the potential information content of the sampled sets of sequences using multiple alignment of complete sequences as the main validation tool. The study considered two criteria: the total number of sequences in BlastP and their associated E-values. The subsequent analyses investigated the influence of the sampling methods on the E-value distributions, the sequence coverage, the final multiple alignment quality and the active site characterisation at various residue conservation thresholds as a function of these criteria.
Conclusion: The comparative analysis of the four sampling methods allows us to propose a suitable sampling strategy that significantly reduces the number of homologous sequences required for alignment, while at the same time maintaining the relevant information concerning the active site residues.
Figures
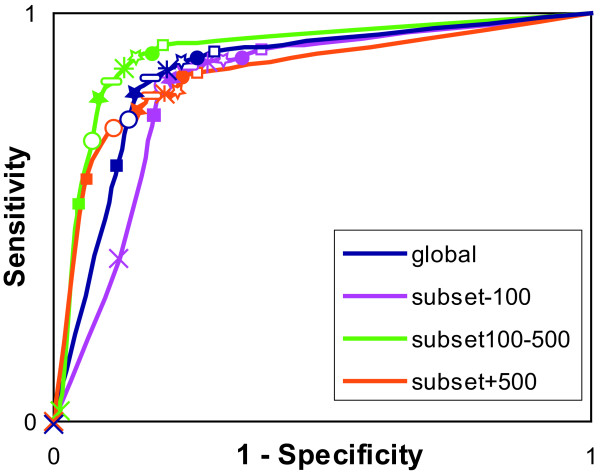
 85%;
85%;  80%;
80%;  75%;
75%;  70%; ● 65%; □ 60%.
70%; ● 65%; □ 60%.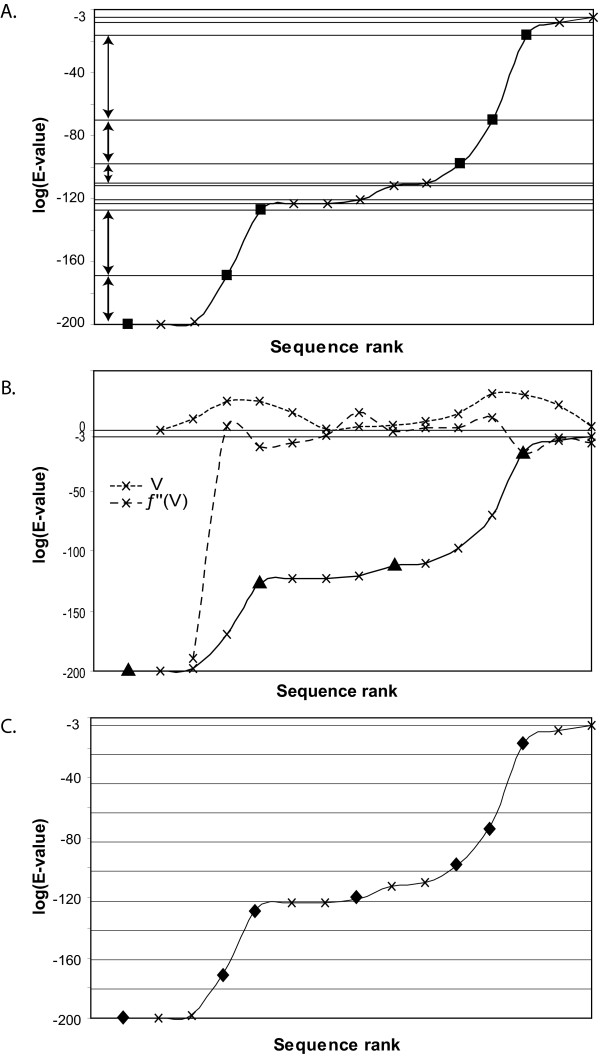
References
-
- Genome OnLine Database http://www.genomesonline.org/
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
