

Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability
- PMID: 17322384
- PMCID: PMC1864880
- DOI: 10.2353/ajpath.2007.060583
Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability
Abstract
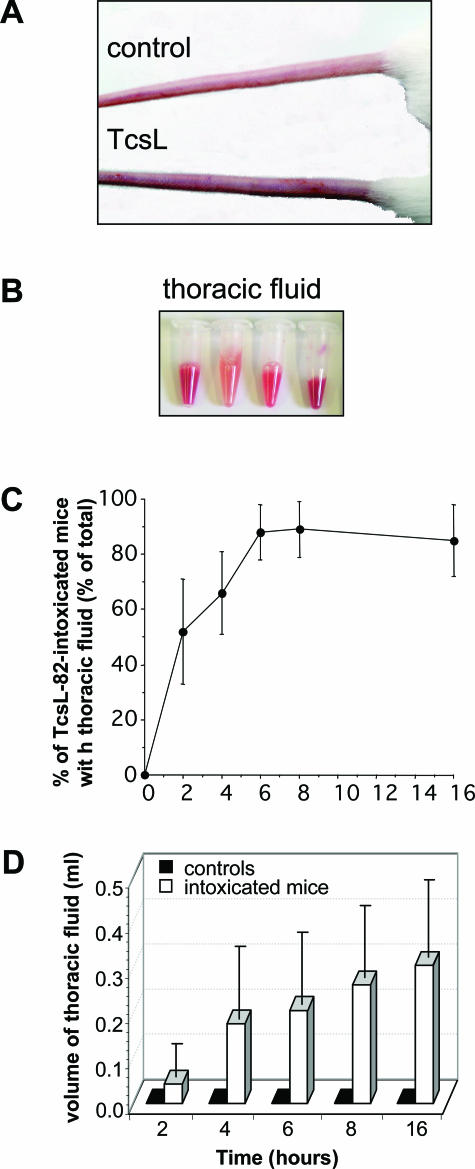
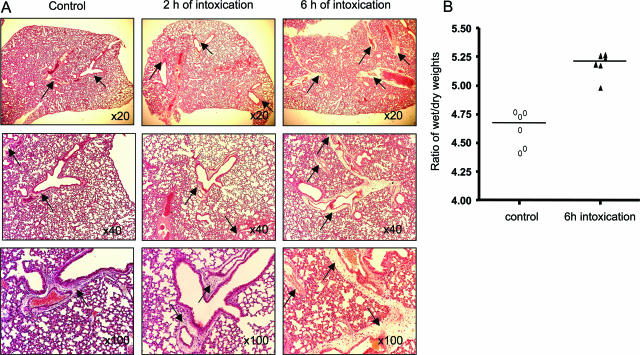
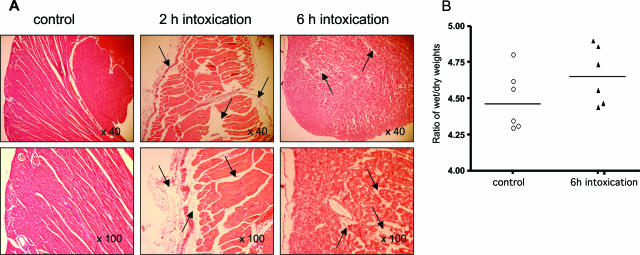
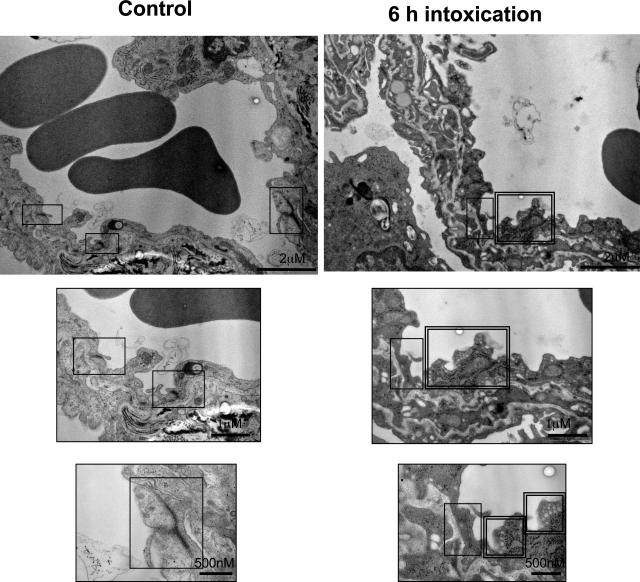
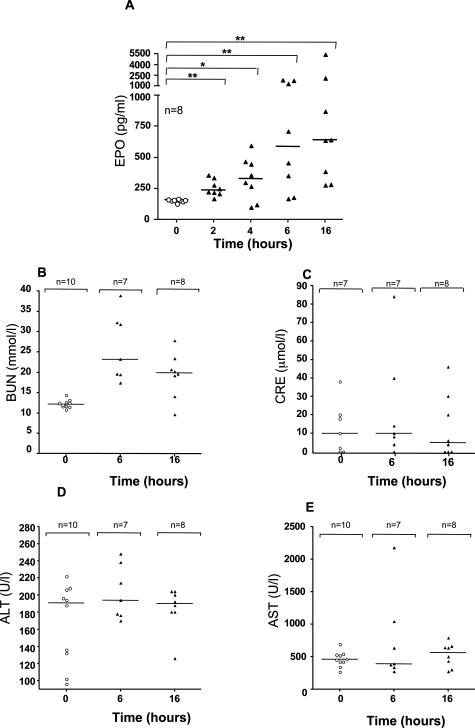
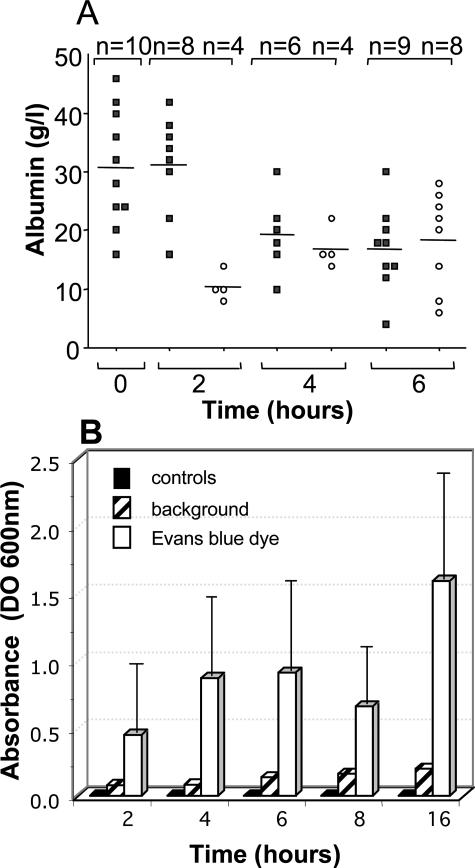
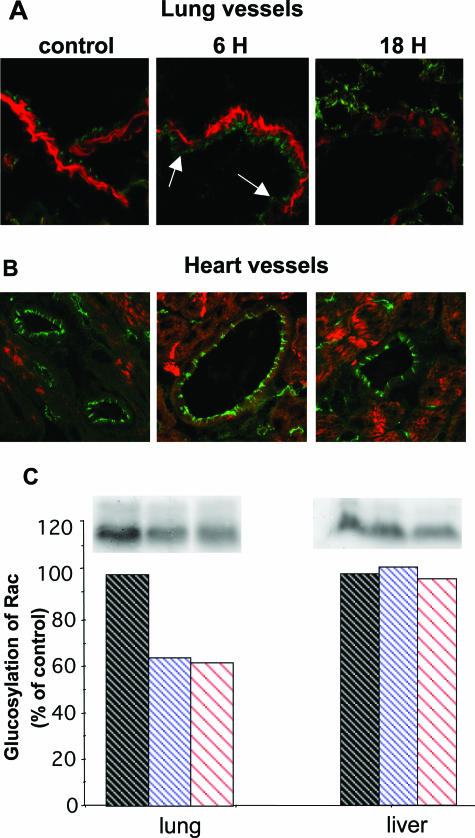
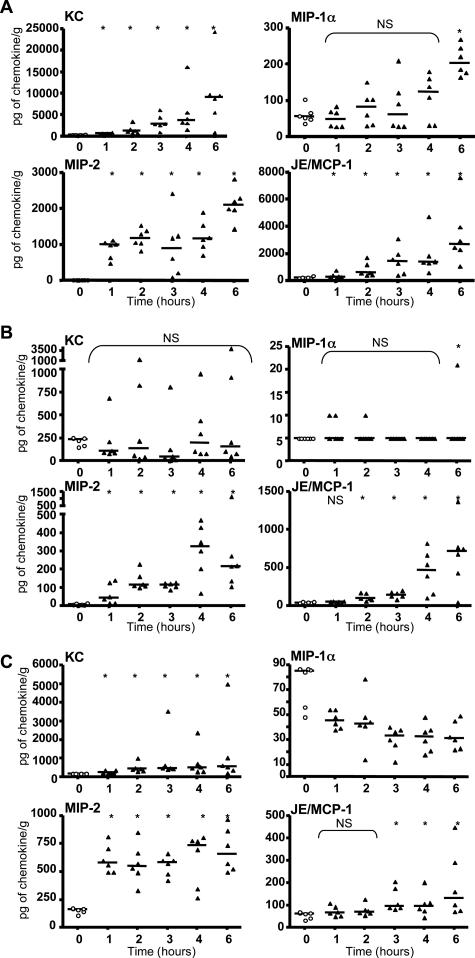
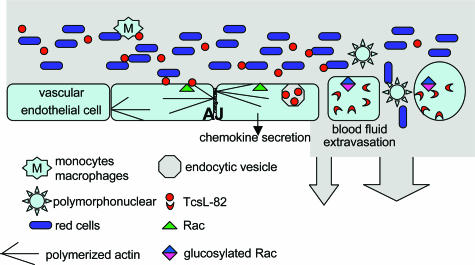
When intraperitoneally injected into Swiss mice, Clostridium sordellii lethal toxin reproduces the fatal toxic shock syndrome observed in humans and animals after natural infection. This animal model was used to study the mechanism of lethal toxin-induced death. Histopathological and biochemical analyses identified lung and heart as preferential organs targeted by lethal toxin. Massive extravasation of blood fluid in the thoracic cage, resulting from an increase in lung vascular permeability, generated profound modifications such as animal dehydration, increase in hematocrit, hypoxia, and finally, cardiorespiratory failure. Vascular permeability increase induced by lethal toxin resulted from modifications of lung endothelial cells as evidenced by electron microscopy. Immunohistochemical analysis demonstrated that VE-cadherin, a protein participating in intercellular adherens junctions, was redistributed from membrane to cytosol in lung endothelial cells. No major sign of lethal toxin-induced inflammation was observed that could participate in the toxic shock syndrome. The main effect of the lethal toxin is the glucosylation-dependent inactivation of small GTPases, in particular Rac, which is involved in actin polymerization occurring in vivo in lungs leading to E-cadherin junction destabilization. We conclude that the cells most susceptible to lethal toxin are lung vascular endothelial cells, the adherens junctions of which were altered after intoxication.
Figures
References
-
- Cunniffe JG. Clostridium sordellii bacteraemia. J Infect. 1996;33:127–129. - PubMed
-
- Kimura AC, Higa JI, Simpson G, Vargas Y, Vugia DJ. Outbreak of necrotizing fascillitis due to Clostridium sordellii among black-tar heroin users. Clin Infect Dis. 2004;38:e87–e89. - PubMed
-
- Fischer M, Bhatnagar J, Guarner J, Reagan S, Hacker JK, Van Meter SH, Poukens V, Whiteman DB, Iton A, Cheung M, Dassey DE, Shieh WJ, Zaki SR. Fatal toxic shock syndrome associated with Clostridium sordellii after medical abortion. N Engl J Med. 2005;353:2352–2360. - PubMed
-
- Miech RP. Pathophysiology of mifepristone-induced septic shock due to Clostridium sordellii. Ann Pharmacother. 2005;39:1483–1488. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
