

PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages
- PMID: 17325208
- PMCID: PMC2064025
- DOI: 10.1083/jcb.200605038
PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages
Abstract
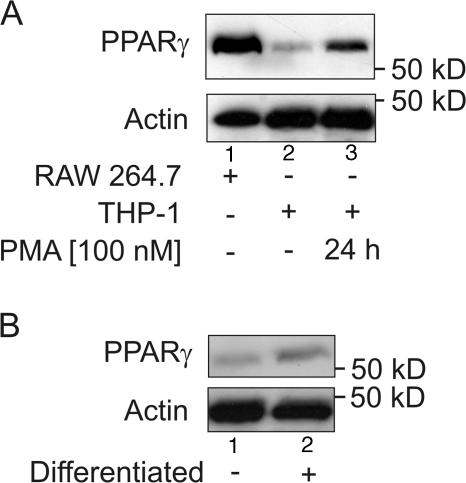
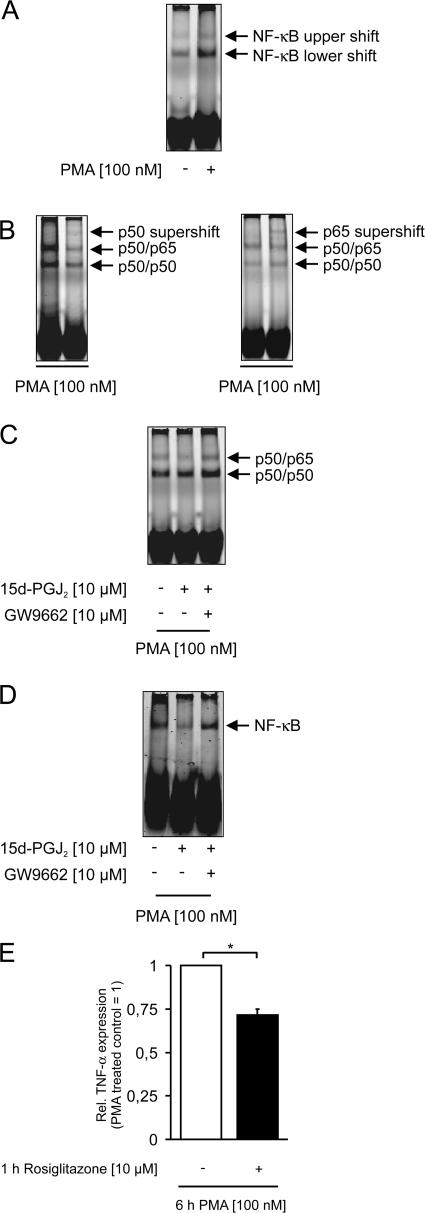
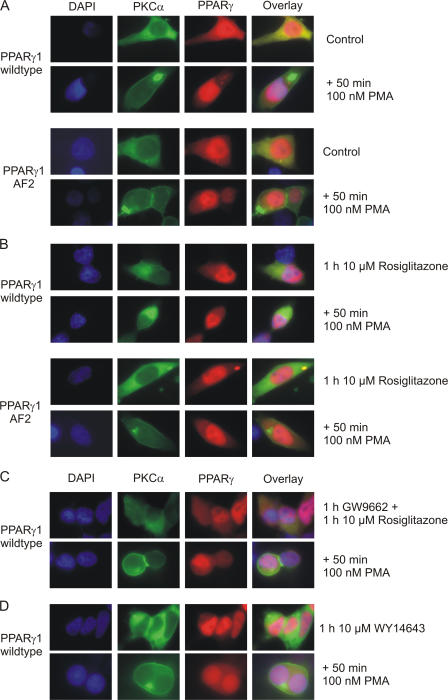
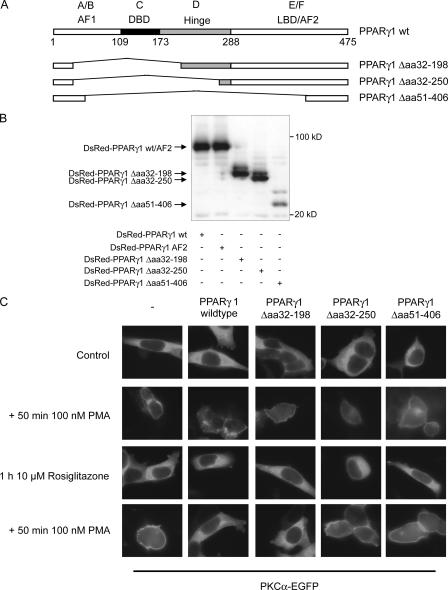
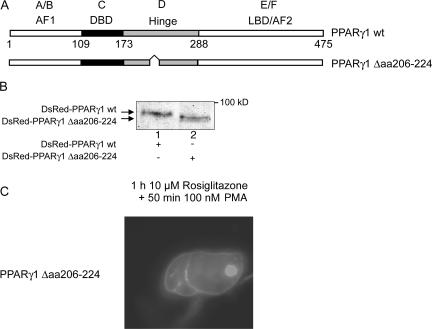
Recently, we provided evidence that PKCalpha depletion in monocytes/macrophages contributes to cellular desensitization during sepsis. We demonstrate that peroxisome proliferator-activated receptor gamma (PPARgamma) agonists dose dependently block PKCalpha depletion in response to the diacylglycerol homologue PMA in RAW 264.7 and human monocyte-derived macrophages. In these cells, we observed PPARgamma-dependent inhibition of nuclear factor-kappaB (NF-kappaB) activation and TNF-alpha expression in response to PMA. Elucidating the underlying mechanism, we found PPARgamma1 expression not only in the nucleus but also in the cytoplasm. Activation of PPARgamma1 wild type, but not an agonist-binding mutant of PPARgamma1, attenuated PMA-mediated PKCalpha cytosol to membrane translocation. Coimmunoprecipitation assays pointed to a protein-protein interaction of PKCalpha and PPARgamma1, which was further substantiated using a mammalian two-hybrid system. Applying PPARgamma1 mutation and deletion constructs, we identified the hinge helix 1 domain of PPARgamma1 that is responsible for PKCalpha binding. Therefore, we conclude that PPARgamma1-dependent inhibition of PKCalpha translocation implies a new model of macrophage desensitization.
Figures





Similar articles
-
PKCalpha: a versatile key for decoding the cellular calcium toolkit.J Cell Biol. 2006 Aug 14;174(4):521-33. doi: 10.1083/jcb.200604033. Epub 2006 Aug 7. J Cell Biol. 2006. PMID: 16893971 Free PMC article.
-
Casein-kinase-II-dependent phosphorylation of PPARgamma provokes CRM1-mediated shuttling of PPARgamma from the nucleus to the cytosol.J Cell Sci. 2010 Jan 15;123(Pt 2):192-201. doi: 10.1242/jcs.055475. Epub 2009 Dec 21. J Cell Sci. 2010. PMID: 20026644
-
PPARgamma agonists inhibit angiogenesis by suppressing PKCalpha- and CREB-mediated COX-2 expression in the human endothelium.Cardiovasc Res. 2010 May 1;86(2):302-10. doi: 10.1093/cvr/cvp400. Epub 2009 Dec 23. Cardiovasc Res. 2010. PMID: 20032081
-
The essentiality of PKCalpha and PKCbetaI translocation for CD14+monocyte differentiation towards macrophages and dendritic cells, respectively.J Cell Biochem. 2007 Oct 1;102(2):429-41. doi: 10.1002/jcb.21305. J Cell Biochem. 2007. PMID: 17455194
-
Peroxisome proliferator-activated receptor gamma1 expression is diminished in human osteoarthritic cartilage and is downregulated by interleukin-1beta in articular chondrocytes.Arthritis Res Ther. 2007;9(2):R31. doi: 10.1186/ar2151. Arthritis Res Ther. 2007. PMID: 17386086 Free PMC article.
Cited by
-
Rescue of neurons from ischemic injury by peroxisome proliferator-activated receptor-gamma requires a novel essential cofactor LMO4.J Neurosci. 2008 Nov 19;28(47):12433-44. doi: 10.1523/JNEUROSCI.2897-08.2008. J Neurosci. 2008. PMID: 19020036 Free PMC article.
-
Breaking the mold: transcription factors in the anucleate platelet and platelet-derived microparticles.Front Immunol. 2015 Feb 13;6:48. doi: 10.3389/fimmu.2015.00048. eCollection 2015. Front Immunol. 2015. PMID: 25762994 Free PMC article. Review.
-
PPARγ is an E3 ligase that induces the degradation of NFκB/p65.Nat Commun. 2012;3:1300. doi: 10.1038/ncomms2270. Nat Commun. 2012. PMID: 23250430
-
PPARgamma1 and LXRalpha face a new regulator of macrophage cholesterol homeostasis and inflammatory responsiveness, AEBP1.Nucl Recept Signal. 2010 Apr 16;8:e004. doi: 10.1621/nrs.08004. Nucl Recept Signal. 2010. PMID: 20419060 Free PMC article. Review.
-
Epithelial Cholesterol Deficiency Attenuates Human Antigen R-linked Pro-inflammatory Stimulation via an SREBP2-linked Circuit.J Biol Chem. 2016 Nov 18;291(47):24641-24656. doi: 10.1074/jbc.M116.723973. Epub 2016 Oct 4. J Biol Chem. 2016. PMID: 27703009 Free PMC article.
References
-
- Abdelrahman, M., A. Sivarajah, and C. Thiemermann. 2005. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovasc. Res. 65:772–781. - PubMed
-
- Abella, A., P. Dubus, M. Malumbres, S.G. Rane, H. Kiyokawa, A. Sicard, F. Vignon, D. Langin, M. Barbacid, and L. Fajas. 2005. Cdk4 promotes adipogenesis through PPARgamma activation. Cell Metab. 2:239–249. - PubMed
-
- Adida, A., and F. Spener. 2006. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. Biochim. Biophys. Acta. 1761:172–181. - PubMed
-
- Akaike, M., W. Che, N.L. Marmarosh, S. Ohta, M. Osawa, B. Ding, B.C. Berk, C. Yan, and J. Abe. 2004. The hinge-helix 1 region of peroxisome proliferator-activated receptor gamma1 (PPARgamma1) mediates interaction with extracellular signal-regulated kinase 5 and PPARgamma1 transcriptional activation: involvement in flow-induced PPARgamma activation in endothelial cells. Mol. Cell. Biol. 24:8691–8704. - PMC - PubMed
-
- Akiyama, T.E., C.T. Baumann, S. Sakai, G.L. Hager, and F.J. Gonzalez. 2002. Selective intranuclear redistribution of PPAR isoforms by RXR alpha. Mol. Endocrinol. 16:707–721. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
