

Kernel-imbedded Gaussian processes for disease classification using microarray gene expression data
- PMID: 17328811
- PMCID: PMC1821044
- DOI: 10.1186/1471-2105-8-67
Kernel-imbedded Gaussian processes for disease classification using microarray gene expression data
Abstract
Background: Designing appropriate machine learning methods for identifying genes that have a significant discriminating power for disease outcomes has become more and more important for our understanding of diseases at genomic level. Although many machine learning methods have been developed and applied to the area of microarray gene expression data analysis, the majority of them are based on linear models, which however are not necessarily appropriate for the underlying connection between the target disease and its associated explanatory genes. Linear model based methods usually also bring in false positive significant features more easily. Furthermore, linear model based algorithms often involve calculating the inverse of a matrix that is possibly singular when the number of potentially important genes is relatively large. This leads to problems of numerical instability. To overcome these limitations, a few non-linear methods have recently been introduced to the area. Many of the existing non-linear methods have a couple of critical problems, the model selection problem and the model parameter tuning problem, that remain unsolved or even untouched. In general, a unified framework that allows model parameters of both linear and non-linear models to be easily tuned is always preferred in real-world applications. Kernel-induced learning methods form a class of approaches that show promising potentials to achieve this goal.
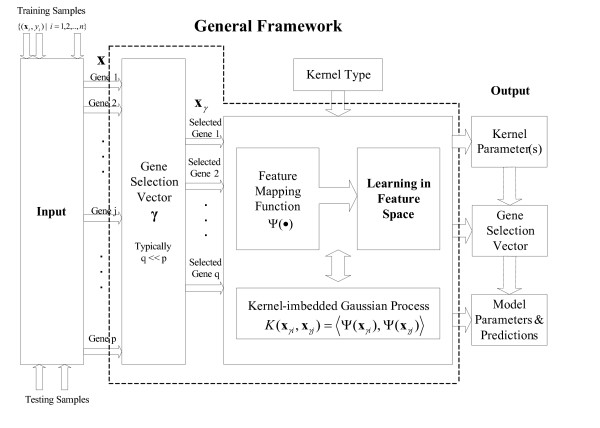
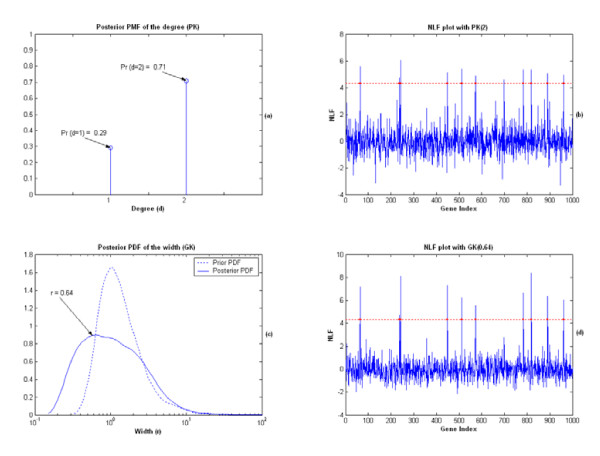
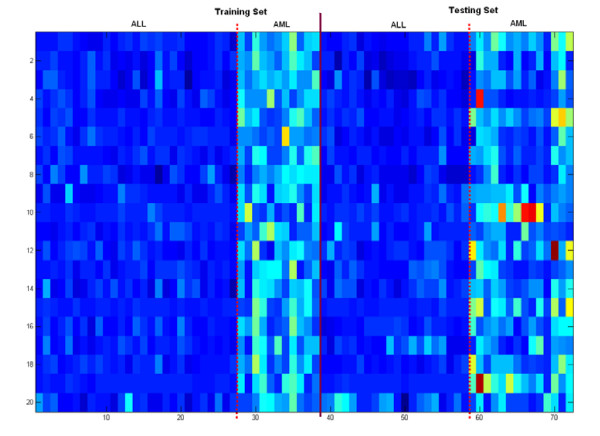
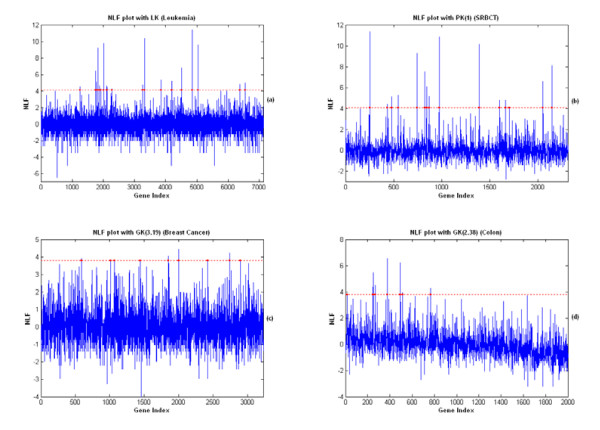
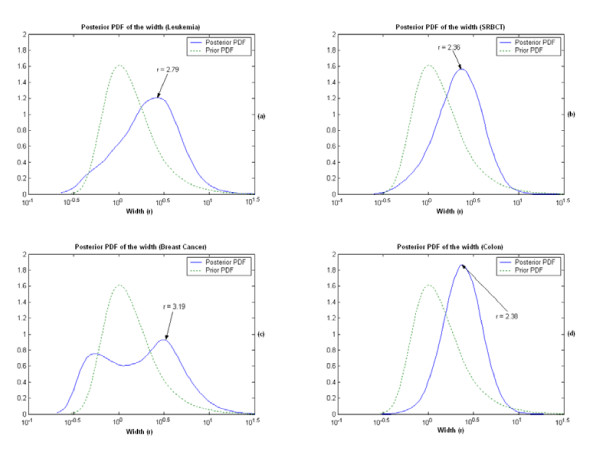
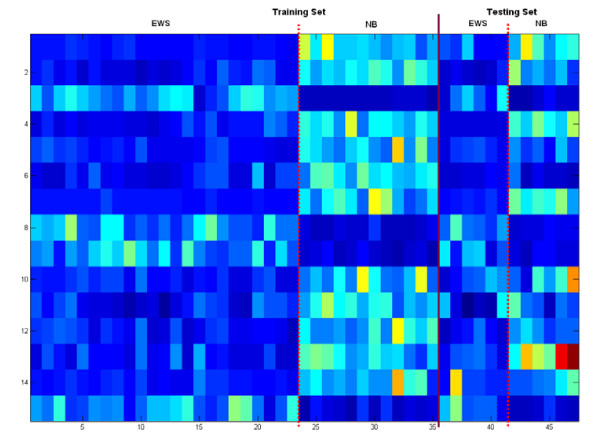
Results: A hierarchical statistical model named kernel-imbedded Gaussian process (KIGP) is developed under a unified Bayesian framework for binary disease classification problems using microarray gene expression data. In particular, based on a probit regression setting, an adaptive algorithm with a cascading structure is designed to find the appropriate kernel, to discover the potentially significant genes, and to make the optimal class prediction accordingly. A Gibbs sampler is built as the core of the algorithm to make Bayesian inferences. Simulation studies showed that, even without any knowledge of the underlying generative model, the KIGP performed very close to the theoretical Bayesian bound not only in the case with a linear Bayesian classifier but also in the case with a very non-linear Bayesian classifier. This sheds light on its broader usability to microarray data analysis problems, especially to those that linear methods work awkwardly. The KIGP was also applied to four published microarray datasets, and the results showed that the KIGP performed better than or at least as well as any of the referred state-of-the-art methods did in all of these cases.
Conclusion: Mathematically built on the kernel-induced feature space concept under a Bayesian framework, the KIGP method presented in this paper provides a unified machine learning approach to explore both the linear and the possibly non-linear underlying relationship between the target features of a given binary disease classification problem and the related explanatory gene expression data. More importantly, it incorporates the model parameter tuning into the framework. The model selection problem is addressed in the form of selecting a proper kernel type. The KIGP method also gives Bayesian probabilistic predictions for disease classification. These properties and features are beneficial to most real-world applications. The algorithm is naturally robust in numerical computation. The simulation studies and the published data studies demonstrated that the proposed KIGP performs satisfactorily and consistently.
Figures
References
-
- Golub TR, Slonim D, Tamayo P, Huard C, Gaasenbeek M, Mesirov J, Coller H, Loh M, Downing J, Caligiuri M, Bloomfield C, Lender E. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. - DOI - PubMed
-
- Ramaswamy S, Golub TR. DNA Microarrays in Clinical Oncology. Journal of Clinical Oncology. 2002;20:1932–1941. - PubMed
-
- Tamayo P, Ramaswamy S. In: "Cancer Genomics and Molecular Pattern Recognition" in Expressing profiling of human tumors: diagnostic and research applications. Marc Ladanyi, William Gerald, editor. Human Press; 2003.
-
- Mallows CL. Some comments on Cp. Technometrics. 1973;15:661–676. doi: 10.2307/1267380. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
