

Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart
- PMID: 17332439
- PMCID: PMC4115784
- DOI: 10.1161/01.RES.0000258451.44949.d7
Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart
Abstract
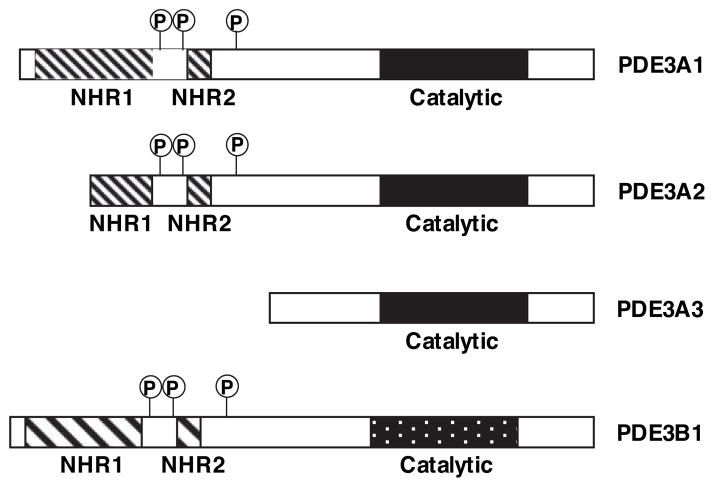
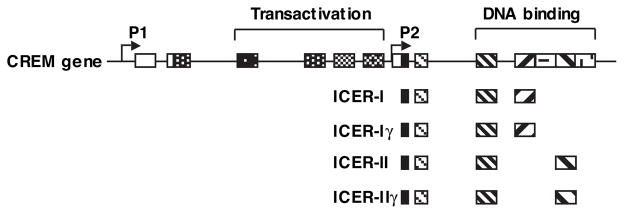
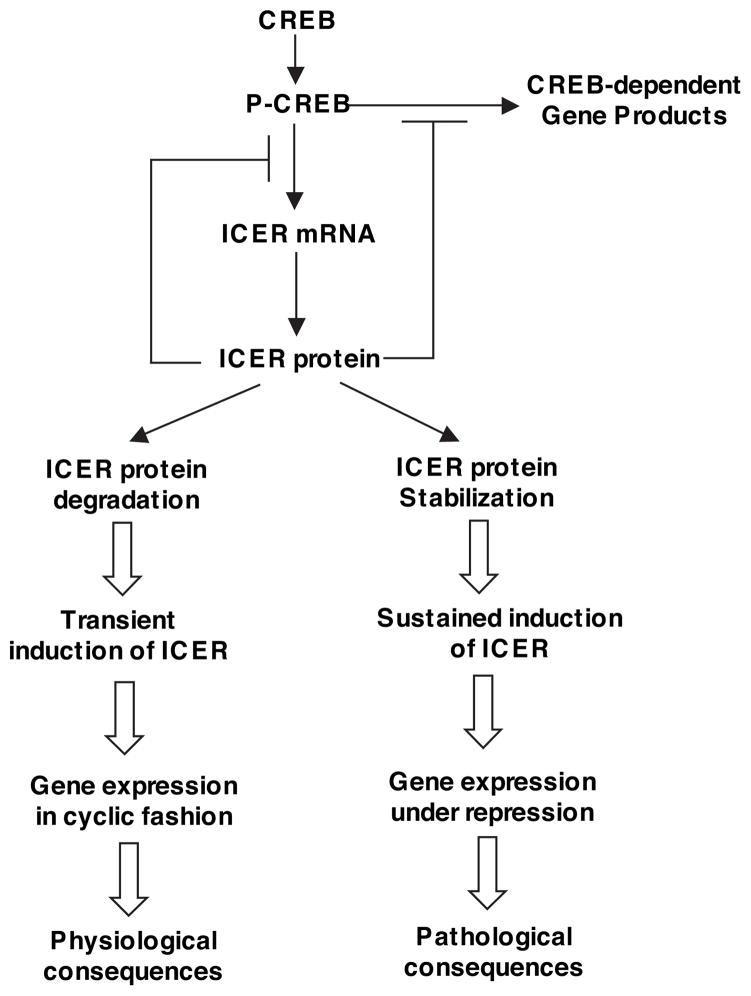
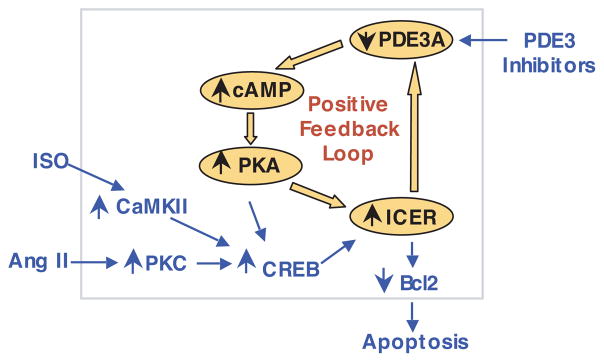
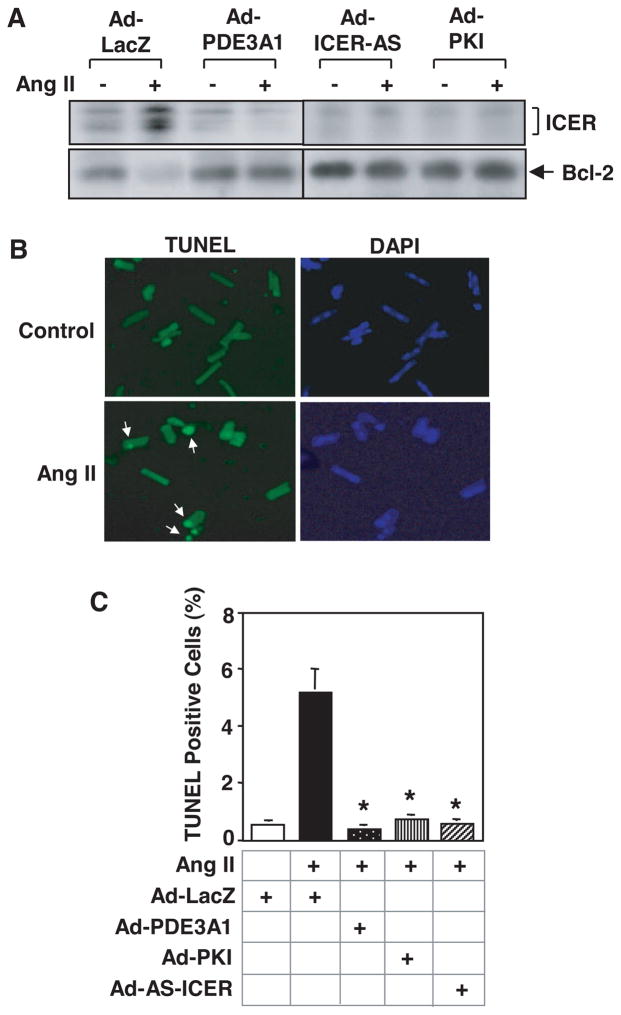
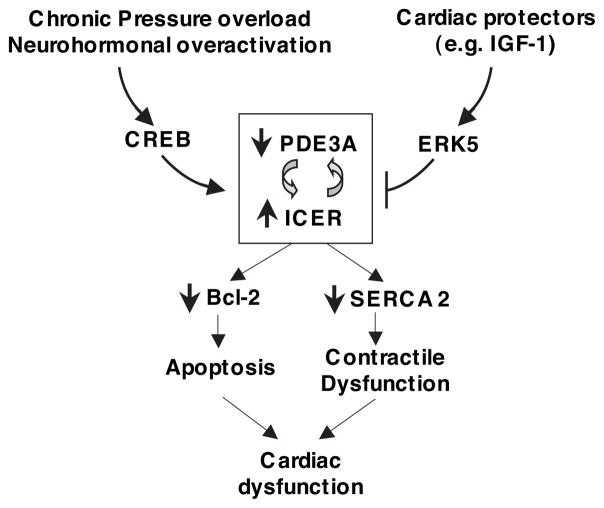
Growing evidence suggests that multiple spatially, temporally, and functionally distinct pools of cyclic nucleotides exist and regulate cardiac performance, from acute myocardial contractility to chronic gene expression and cardiac structural remodeling. Cyclic nucleotide phosphodiesterases (PDEs), by hydrolyzing cAMP and cyclic GMP, regulate the amplitude, duration, and compartmentation of cyclic nucleotide-mediated signaling. In particular, PDE3 enzymes play a major role in regulating cAMP metabolism in the cardiovascular system. PDE3 inhibitors, by raising cAMP content, have acute inotropic and vasodilatory effects in treating congestive heart failure but have increased mortality in long-term therapy. PDE3A expression is downregulated in human and animal failing hearts. In vitro, inhibition of PDE3A function is associated with myocyte apoptosis through sustained induction of a transcriptional repressor ICER (inducible cAMP early repressor) and thereby inhibition of antiapoptotic molecule Bcl-2 expression. Sustained induction of ICER may also cause the change of other protein expression implicated in human and animal failing hearts. These data suggest that the downregulation of PDE3A observed in failing hearts may play a causative role in the progression of heart failure, in part, by inducing ICER and promoting cardiac myocyte dysfunction. Hence, strategies that maintain PDE3A function may represent an attractive approach to circumvent myocyte apoptosis and cardiac dysfunction.
Figures
References
-
- Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. - PubMed
-
- Lohmann SM, Fischmeister R, Walter U. Signal transduction by cGMP in heart. Basic Res Cardiol. 1991;86:503–514. - PubMed
-
- Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr Opin Cell Biol. 2000;12:174–179. - PubMed
-
- Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
