

ECM remodeling in hypertensive heart disease
- PMID: 17332884
- PMCID: PMC1804378
- DOI: 10.1172/JCI31044
ECM remodeling in hypertensive heart disease
Abstract
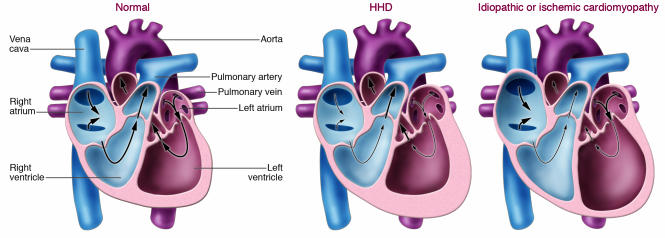
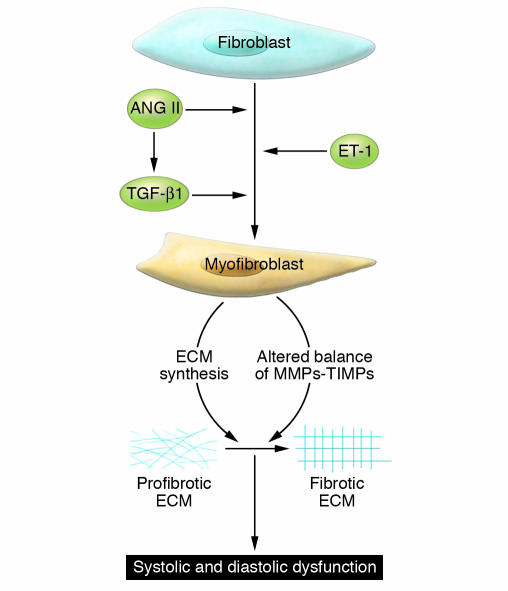
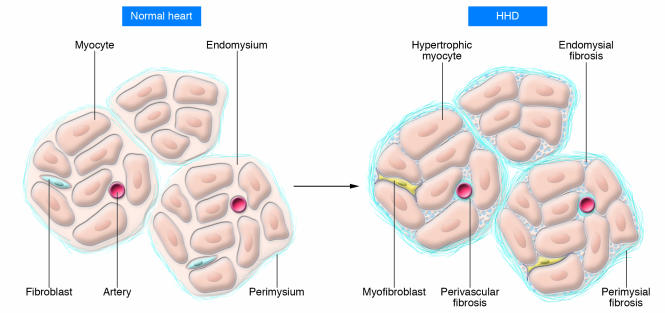
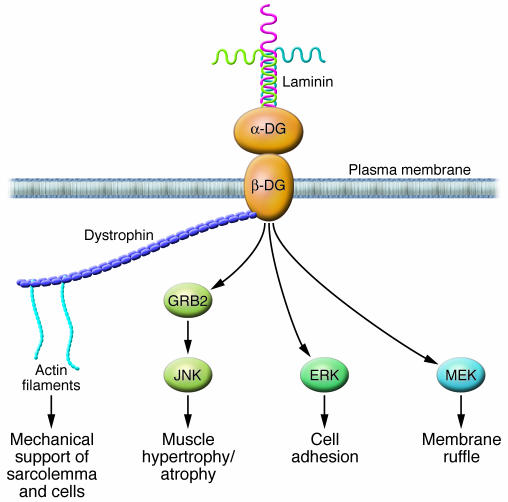
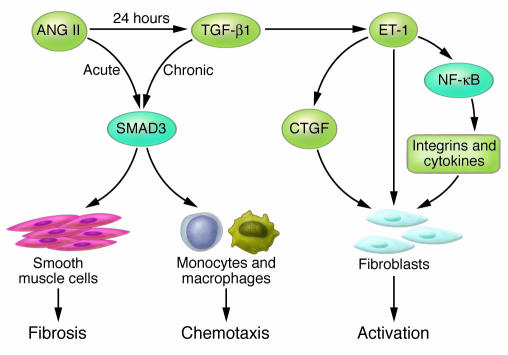
Hypertensive heart disease (HHD) occurs in patients that clinically have both diastolic and systolic heart failure and will soon become the most common cause of heart failure. Two key aspects of heart failure secondary to HHD are the relatively highly prevalent LV hypertrophy and cardiac fibrosis, caused by changes in the local and systemic neurohormonal environment. The fibrotic state is marked by changes in the balance between MMPs and their inhibitors, which alter the composition of the ECM. Importantly, the fibrotic ECM impairs cardiomyocyte function. Recent research suggests that therapies targeting the expression, synthesis, or activation of the enzymes responsible for ECM homeostasis might represent novel opportunities to modify the natural progression of HHD.
Figures
References
-
- Zile M.R., Brutsaert D.L. New concepts in diastolic dysfunction and diastolic heart failure: part II: causal mechanisms and treatment. Circulation. 2002;105:1503–1508. - PubMed
-
- Zile M.R., Brutsaert D.L. New concepts in diastolic dysfunction and diastolic heart failure: part I: diagnosis, prognosis, and measurements of diastolic function. Circulation. 2002;105:1387–1393. - PubMed
-
- Shirwany A., Weber K.T. Extracellular matrix remodeling in hypertensive heart disease. . J. Am. Coll. Cardiol. 2006;48:97–98. - PubMed
-
- Gandhi S.K., et al. The pathogenesis of acute pulmonary edema associated with hypertension. . N. Engl. J. Med. 2001;344:17–22. - PubMed
-
- Benjamin E.J., Levy D. Why is left ventricular hypertrophy so predictive of morbidity and mortality? Am. J. Med. Sci. 1999;317:168–175. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
