

Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases
- PMID: 17338813
- PMCID: PMC1829167
- DOI: 10.1186/1471-2105-8-73
Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases
Abstract
Background: SPOUT methyltransferases (MTases) are a large class of S-adenosyl-L-methionine-dependent enzymes that exhibit an unusual alpha/beta fold with a very deep topological knot. In 2001, when no crystal structures were available for any of these proteins, Anantharaman, Koonin, and Aravind identified homology between SpoU and TrmD MTases and defined the SPOUT superfamily. Since then, multiple crystal structures of knotted MTases have been solved and numerous new homologous sequences appeared in the databases. However, no comprehensive comparative analysis of these proteins has been carried out to classify them based on structural and evolutionary criteria and to guide functional predictions.
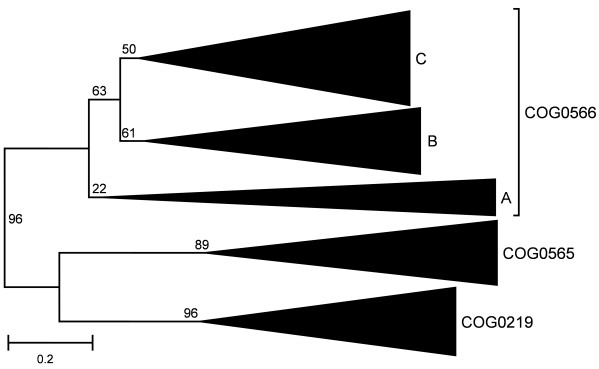
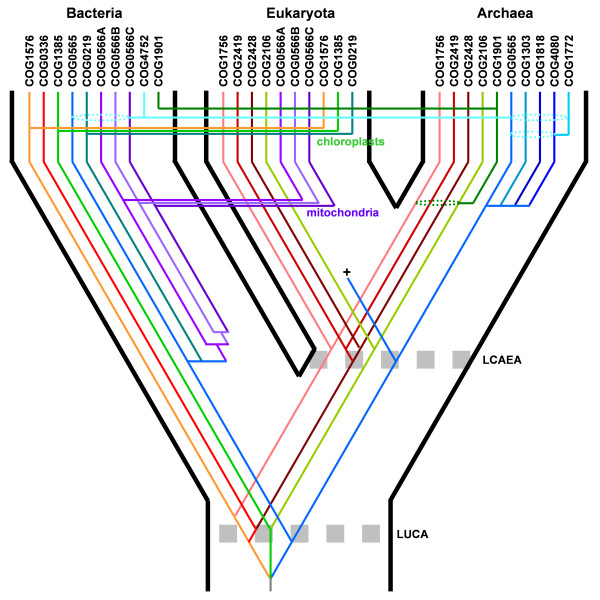
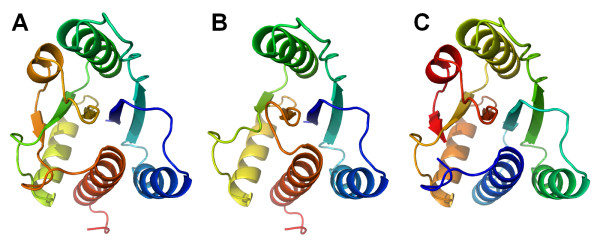
Results: We carried out extensive searches of databases of protein structures and sequences to collect all members of previously identified SPOUT MTases, and to identify previously unknown homologs. Based on sequence clustering, characterization of domain architecture, structure predictions and sequence/structure comparisons, we re-defined families within the SPOUT superfamily and predicted putative active sites and biochemical functions for the so far uncharacterized members. We have also delineated the common core of SPOUT MTases and inferred a multiple sequence alignment for the conserved knot region, from which we calculated the phylogenetic tree of the superfamily. We have also studied phylogenetic distribution of different families, and used this information to infer the evolutionary history of the SPOUT superfamily.
Conclusion: We present the first phylogenetic tree of the SPOUT superfamily since it was defined, together with a new scheme for its classification, and discussion about conservation of sequence and structure in different families, and their functional implications. We identified four protein families as new members of the SPOUT superfamily. Three of these families are functionally uncharacterized (COG1772, COG1901, and COG4080), and one (COG1756 represented by Nep1p) has been already implicated in RNA metabolism, but its biochemical function has been unknown. Based on the inference of orthologous and paralogous relationships between all SPOUT families we propose that the Last Universal Common Ancestor (LUCA) of all extant organisms contained at least three SPOUT members, ancestors of contemporary RNA MTases that carry out m1G, m3U, and 2'O-ribose methylation, respectively. In this work we also speculate on the origin of the knot and propose possible 'unknotted' ancestors. The results of our analysis provide a comprehensive 'roadmap' for experimental characterization of SPOUT MTases and interpretation of functional studies in the light of sequence-structure relationships.
Figures










References
-
- Anantharaman V, Koonin EV, Aravind L. SPOUT: a class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J Mol Microbiol Biotechnol. 2002;4:71–75. - PubMed
-
- Nureki O, Shirouzu M, Hashimoto K, Ishitani R, Terada T, Tamakoshi M, Oshima T, Chijimatsu M, Takio K, Vassylyev DG, Shibata T, Inoue Y, Kuramitsu S, Yokoyama S. An enzyme with a deep trefoil knot for the active-site architecture. Acta Crystallogr D Biol Crystallogr. 2002;58:1129–1137. doi: 10.1107/S0907444902006601. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
