

Genetics and mediators in pulmonary arterial hypertension
- PMID: 17338927
- PMCID: PMC3740514
- DOI: 10.1016/j.ccm.2006.11.007
Genetics and mediators in pulmonary arterial hypertension
Abstract
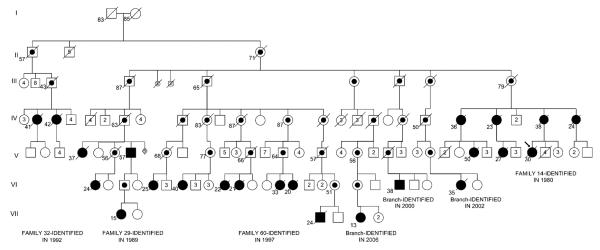
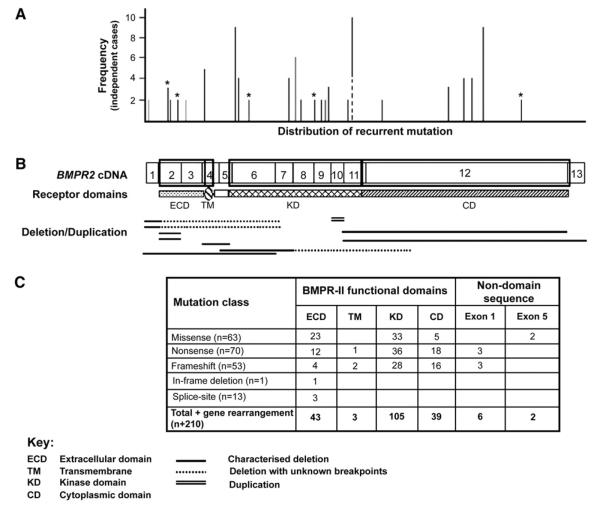
Pulmonary arterial hypertension (PAH) is an uncommon disorder of the pulmonary vasculature characterized by remodeling of the smallest pulmonary arteries, leading to a progressive increase in pulmonary vascular resistance. Various forms of PAH exist, including familial (FPAH) and idiopathic (IPAH) forms and associated conditions. FPAH transmits as an autosomal dominant trait that exhibits genetic anticipation but also markedly reduced penetrance (20%). The primary genetic defect of FPAH, identifiable in more than 70% of cases of FPAH, is a mutation in the gene encoding bone morphogenetic protein receptor type 2 (BMPR2), a member of the transforming growth factor beta superfamily. The true prevalence of BMPR2 mutations in IPAH is unknown, with reports ranging from 10% to 40% of patients. The cause of the variable phenotypic expression of PAH among carriers of mutated BMPR2 genes and patients is unclear, and likely related to environmental and genetic modifiers of disease not yet fully elucidated. Although BMPR2-related pathways seem to be pivotal, many other mediator pathways participate in the pathogenesis of different forms of PAH and are being actively investigated, both independently and in combination. As understanding of the molecular basis of this devastating disease improves, opportunities for earlier diagnosis, additional therapeutic regimens, and perhaps disease prevention will emerge.
Figures
Similar articles
-
Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension.Hum Mutat. 2006 Feb;27(2):121-32. doi: 10.1002/humu.20285. Hum Mutat. 2006. PMID: 16429395
-
Idiopathic pulmonary hypertension: what did we learn from genes?Sarcoidosis Vasc Diffuse Lung Dis. 2005 Dec;22 Suppl 1:S91-100. Sarcoidosis Vasc Diffuse Lung Dis. 2005. PMID: 16457021 Review.
-
A novel mutation in the BMPR2 gene in familial pulmonary arterial hypertension.Chin Med J (Engl). 2008 Mar 5;121(5):399-404. Chin Med J (Engl). 2008. PMID: 18364108
-
Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele.Hum Mutat. 2009 Apr;30(4):649-54. doi: 10.1002/humu.20922. Hum Mutat. 2009. PMID: 19206171 Free PMC article.
-
Genetics and genomics of pulmonary arterial hypertension.J Am Coll Cardiol. 2009 Jun 30;54(1 Suppl):S32-S42. doi: 10.1016/j.jacc.2009.04.015. J Am Coll Cardiol. 2009. PMID: 19555857 Free PMC article. Review.
Cited by
-
Pulmonary arterial hypertension associated with systemic sclerosis.Expert Rev Respir Med. 2011 Apr;5(2):267-79. doi: 10.1586/ers.11.18. Expert Rev Respir Med. 2011. PMID: 21510736 Free PMC article. Review.
-
What patients and their relatives think about testing for BMPR2.J Genet Couns. 2008 Oct;17(5):452-8. doi: 10.1007/s10897-008-9172-1. Epub 2008 Sep 13. J Genet Couns. 2008. PMID: 18791814 Free PMC article.
-
Systemic sclerosis-associated pulmonary arterial hypertension.Chest. 2013 Oct;144(4):1346-1356. doi: 10.1378/chest.12-2396. Chest. 2013. PMID: 24081346 Free PMC article. Review.
-
Pulmonary vascular disease in Gaucher disease: clinical spectrum, determinants of phenotype and long-term outcomes of therapy.J Inherit Metab Dis. 2011 Jun;34(3):643-50. doi: 10.1007/s10545-011-9313-9. Epub 2011 Mar 29. J Inherit Metab Dis. 2011. PMID: 21445609 Free PMC article.
-
An endothelial activin A-bone morphogenetic protein receptor type 2 link is overdriven in pulmonary hypertension.Nat Commun. 2021 Mar 19;12(1):1720. doi: 10.1038/s41467-021-21961-3. Nat Commun. 2021. PMID: 33741934 Free PMC article.
References
-
- Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. 1951;11(6):686–705. - PubMed
-
- Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis. 1984;129(1):194–7. - PubMed
-
- Thomas AQ, Gaddipati R, Newman JH, et al. Genetics of primary pulmonary hypertension. Clin Chest Med. 2001;22(3):477–91. ix. - PubMed
-
- Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107(2):216–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
