

A maximum-likelihood method for the estimation of pairwise relatedness in structured populations
- PMID: 17339212
- PMCID: PMC1893072
- DOI: 10.1534/genetics.106.063149
A maximum-likelihood method for the estimation of pairwise relatedness in structured populations
Abstract
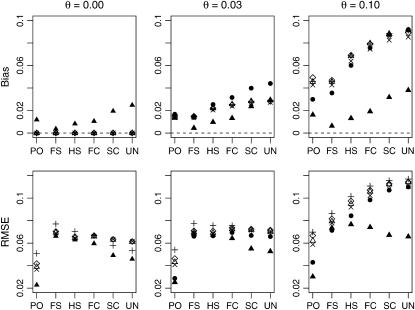
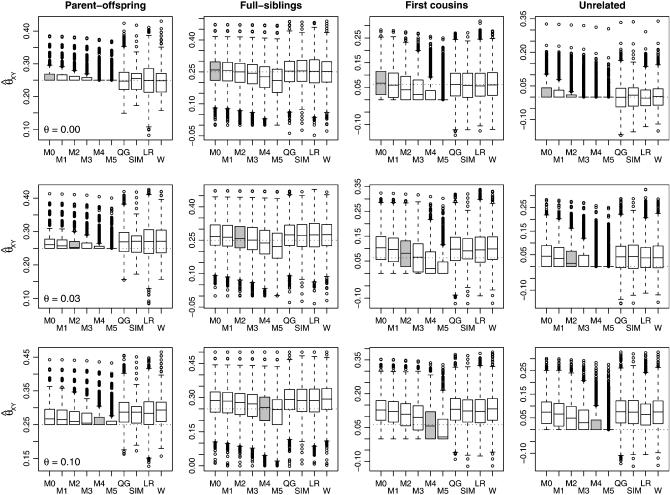
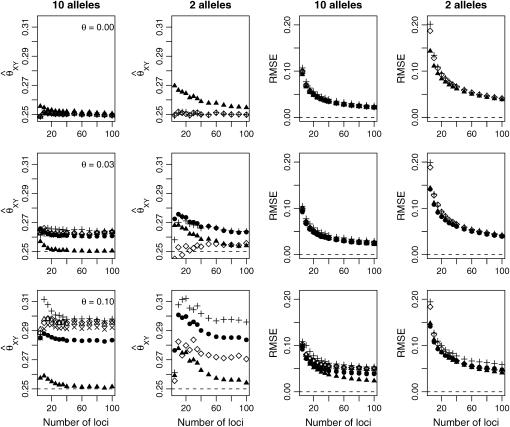
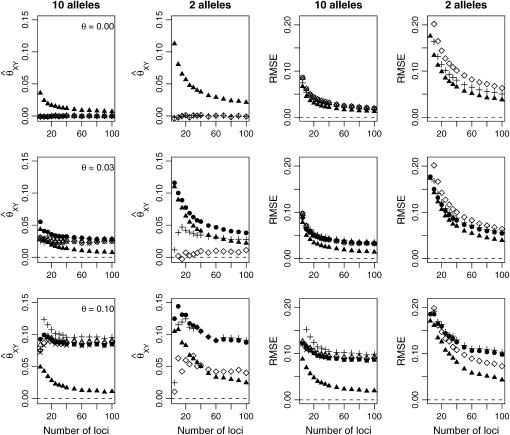
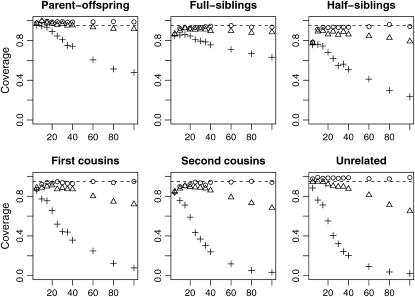
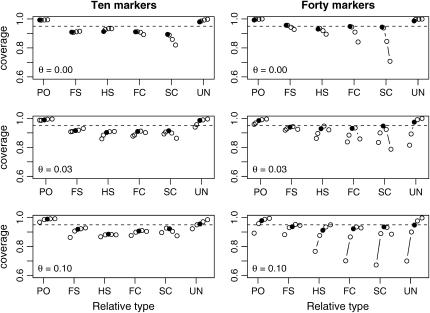
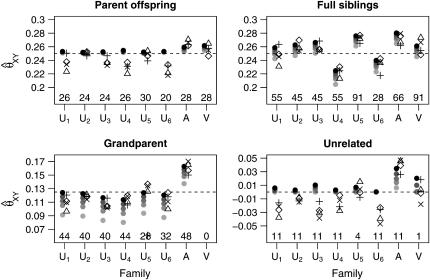
A maximum-likelihood estimator for pairwise relatedness is presented for the situation in which the individuals under consideration come from a large outbred subpopulation of the population for which allele frequencies are known. We demonstrate via simulations that a variety of commonly used estimators that do not take this kind of misspecification of allele frequencies into account will systematically overestimate the degree of relatedness between two individuals from a subpopulation. A maximum-likelihood estimator that includes F(ST) as a parameter is introduced with the goal of producing the relatedness estimates that would have been obtained if the subpopulation allele frequencies had been known. This estimator is shown to work quite well, even when the value of F(ST) is misspecified. Bootstrap confidence intervals are also examined and shown to exhibit close to nominal coverage when F(ST) is correctly specified.
Figures
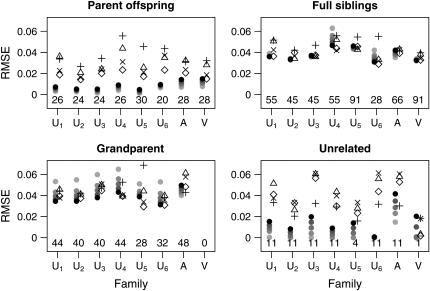
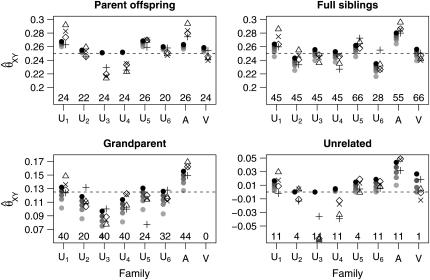
References
-
- Ayres, K. L., 2000. Relatedness testing in subdivided populations. Forensic Sci. Int. 114: 107–115. - PubMed
-
- Balding, D. J., and R. A. Nichols, 1994. DNA profile match probability calculation: how to allow for population stratification, relatedness, database selection and single bands. Forensic Sci. Int. 64: 125–140. - PubMed
-
- Balding, D. J., and R. A. Nichols, 1997. Significant genetic correlations among Caucasians at forensic DNA loci. Heredity 78: 583–589. - PubMed
-
- Budowle, B., and K. L. Monson, 1994. Greater differences in forensic DNA profile frequencies estimated from racial groups than from ethnic subgroups. Clin. Chim. Acta 228: 3–18. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
