

USER friendly DNA engineering and cloning method by uracil excision
- PMID: 17341463
- PMCID: PMC1874603
- DOI: 10.1093/nar/gkm041
USER friendly DNA engineering and cloning method by uracil excision
Abstract
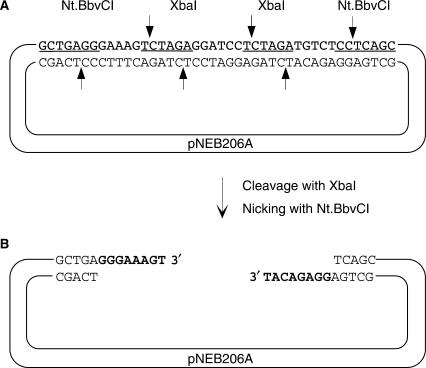
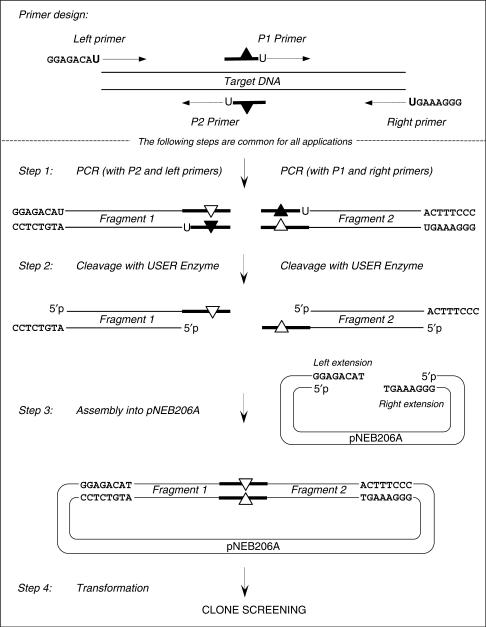
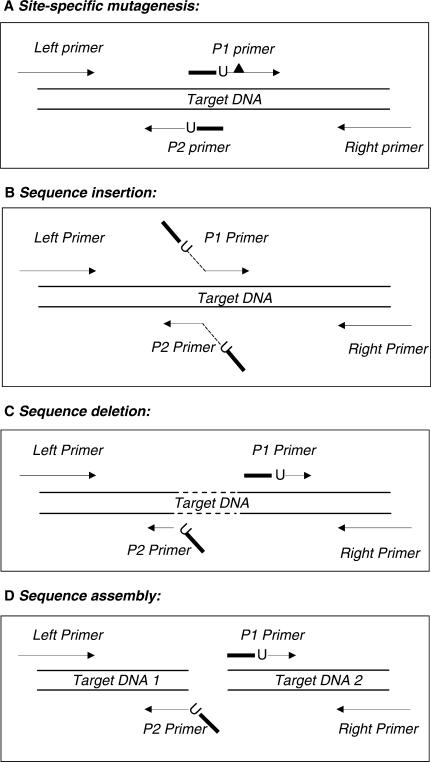
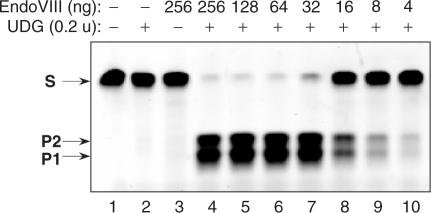
Here we report a PCR-based DNA engineering technique for seamless assembly of recombinant molecules from multiple components. We create cloning vector and target molecules flanked with compatible single-stranded (ss) extensions. The vector contains a cassette with two inversely oriented nicking endonuclease sites separated by restriction endonuclease site(s). The spacer sequences between the nicking and restriction sites are tailored to create ss extensions of custom sequence. The vector is then linearized by digestion with nicking and restriction endonucleases. To generate target molecules, a single deoxyuridine (dU) residue is placed 6-10 nt away from the 5'-end of each PCR primer. 5' of dU the primer sequence is compatible either with an ss extension on the vector or with the ss extension of the next-in-line PCR product. After amplification, the dU is excised from the PCR products with the USER enzyme leaving PCR products flanked by 3' ss extensions. When mixed together, the linearized vector and PCR products directionally assemble into a recombinant molecule through complementary ss extensions. By varying the design of the PCR primers, the protocol is easily adapted to perform one or more simultaneous DNA manipulations such as directional cloning, site-specific mutagenesis, sequence insertion or deletion and sequence assembly.
Figures


References
-
- Hofer B, Kuhlein B. A high efficiency method for site-directed mutagenesis with any plasmid. Gene. 1989;84:153–157. - PubMed
-
- Hashimoto-Gotoh T, Mizuno T, Ogasahara Y, Nakagawa M. An oligodeoxyribonucleotide-directed dual amber method for site-directed mutagenesis. Gene. 1995;152:271–275. - PubMed
-
- Deng WP, Nickoloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 1992;200:81–88. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
