

Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53
- PMID: 17344317
- PMCID: PMC1874625
- DOI: 10.1093/nar/gkm099
Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53
Abstract
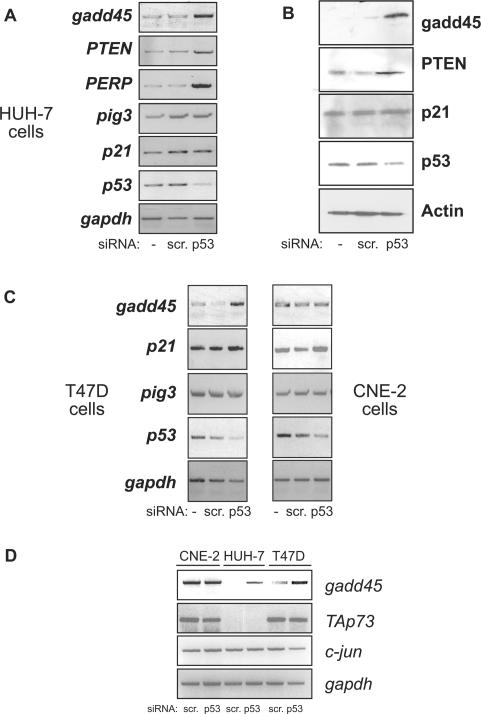
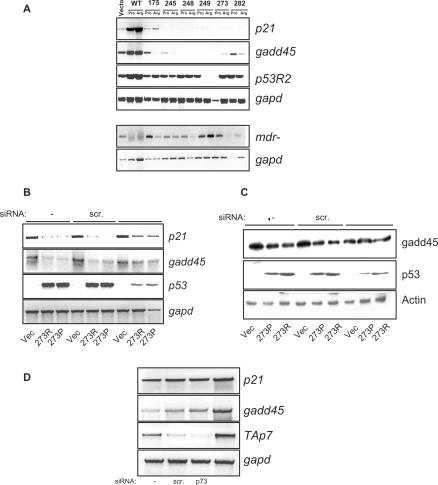
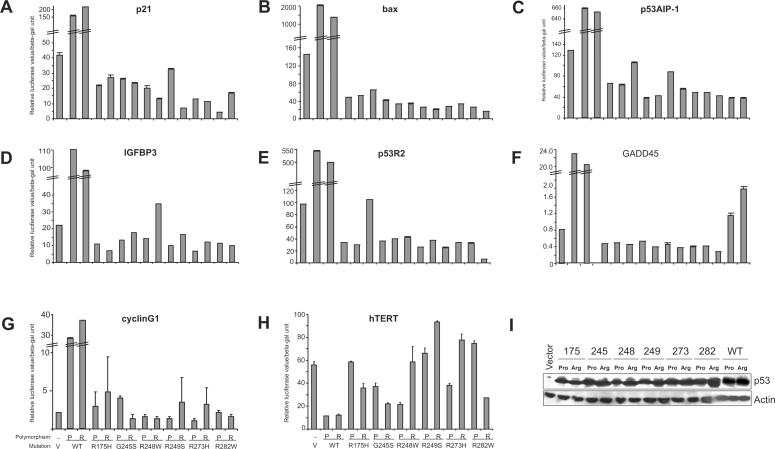
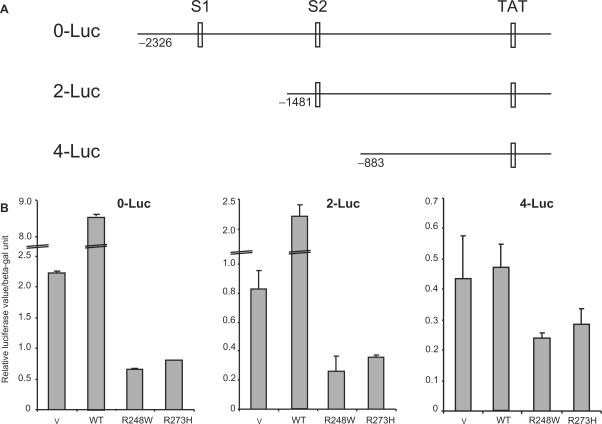
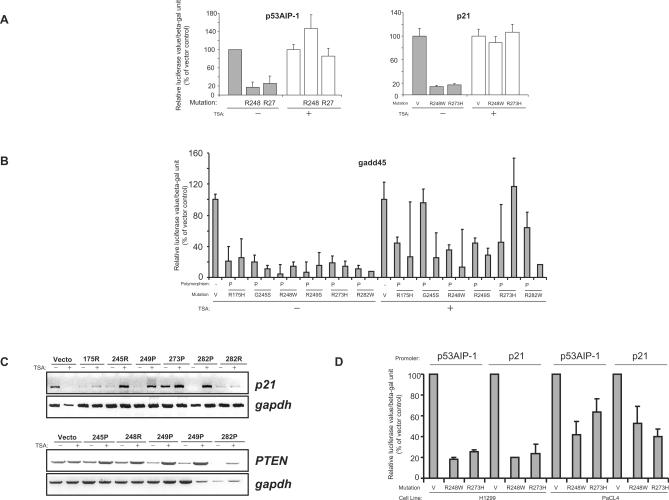
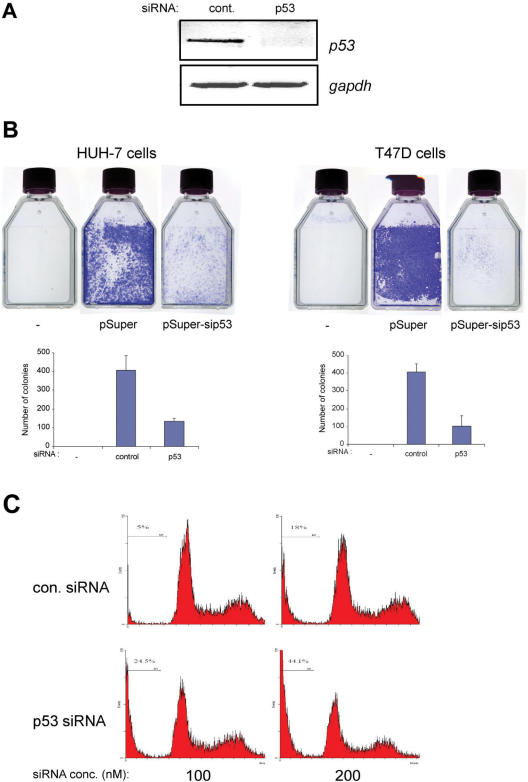
Tumour-derived p53 mutants are thought to have acquired 'gain-of-function' properties that contribute to oncogenicity. We have tested the hypothesis that p53 mutants suppress p53-target gene expression, leading to enhanced cellular growth. Silencing of mutant p53 expression in several human cell lines was found to lead to the upregulation of wild-type p53-target genes such as p21, gadd45, PERP and PTEN. The expression of these genes was also suppressed in H1299-based isogenic cell lines expressing various hot-spot p53 mutants, and silencing of mutant p53, but not TAp73, abrogated the suppression. Consistently, these hot-spot p53 mutants were able to suppress a variety of p53-target gene promoters. Analysis using the proto-type p21 promoter construct indicated that the p53-binding sites are dispensable for mutant p53-mediated suppression. However, treatment with the histone deacetylase inhibitor trichostatin-A resulted in relief of mutant p53-mediated suppression, suggesting that mutant p53 may induce hypo-acetylation of target gene promoters leading to the suppressive effects. Finally, we show that stable down-regulation of mutant p53 expression resulted in reduced cellular colony growth in human cancer cells, which was found to be due to the induction of apoptosis. Together, the results demonstrate another mechanism through which p53 mutants could promote cellular growth.
Figures
Comment in
-
Editor's Note on 'Cancer-derived p53 mutants suppress p53-target gene expression-potential mechanism for gain of function of mutant p53'.Nucleic Acids Res. 2023 Jan 25;51(2):997-1000. doi: 10.1093/nar/gkac1265. Nucleic Acids Res. 2023. PMID: 36625291 Free PMC article. No abstract available.
References
-
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. - PubMed
-
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. - PubMed
-
- Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC. TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ. 2004;157:247–270. - PubMed
-
- Roemer K. Mutant p53: gain-of-function oncoproteins and wild-type p53 inactivators. Biol. Chem. 1999;380:879–887. - PubMed
-
- Vikhanskaya F, Siddique MM, Lee MK, Broggini M, Sabapathy K. Evaluation of the combined effect of p53 codon 72 polymorphism and hotspot mutations in response to anticancer drugs. Clin. Cancer Res. 2005;11:4348–4356. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
