

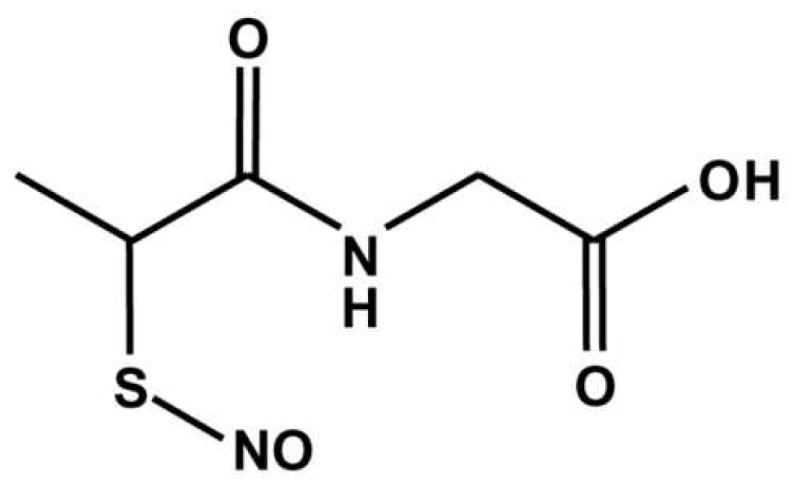
Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury
- PMID: 17350035
- PMCID: PMC2134894
- DOI: 10.1016/j.yjmcc.2007.01.010
Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury
Abstract
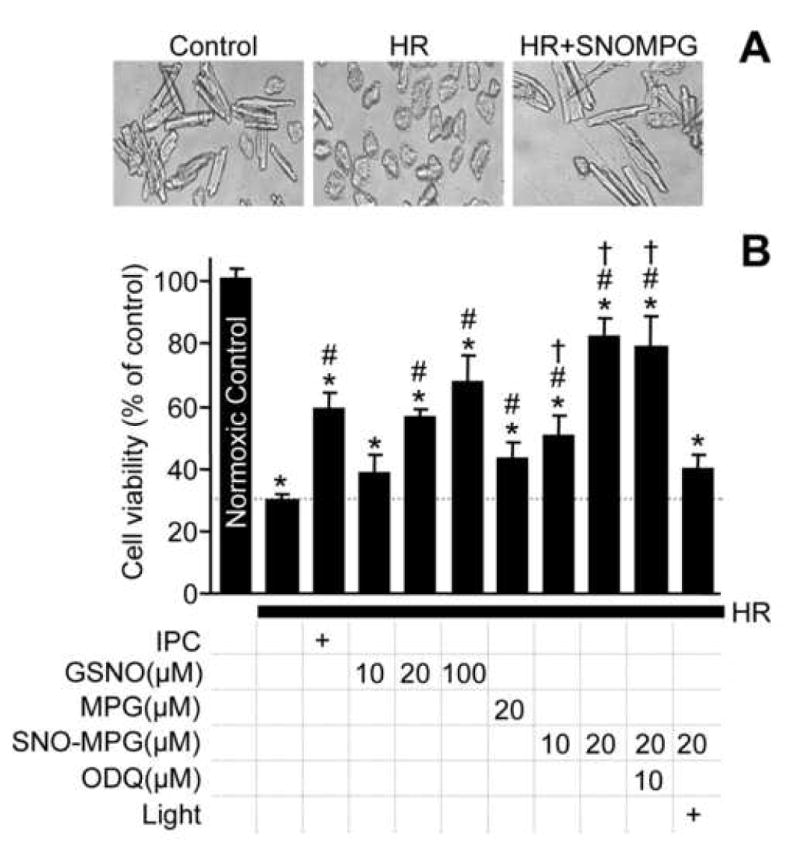
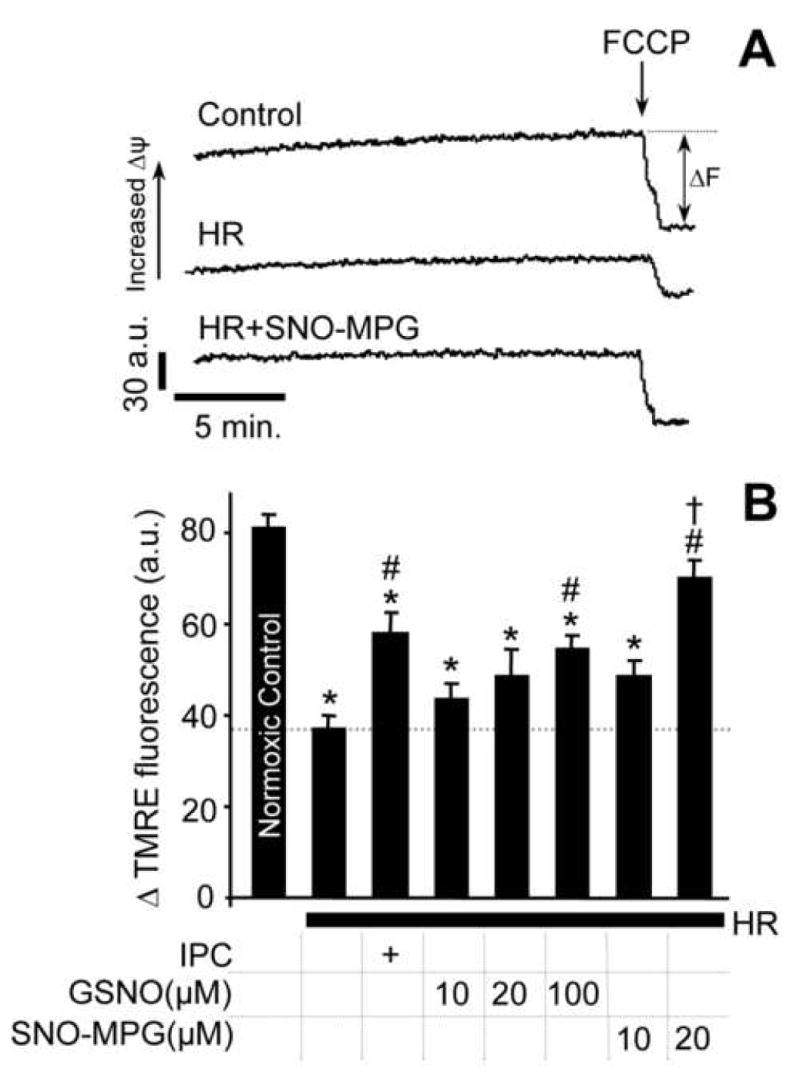
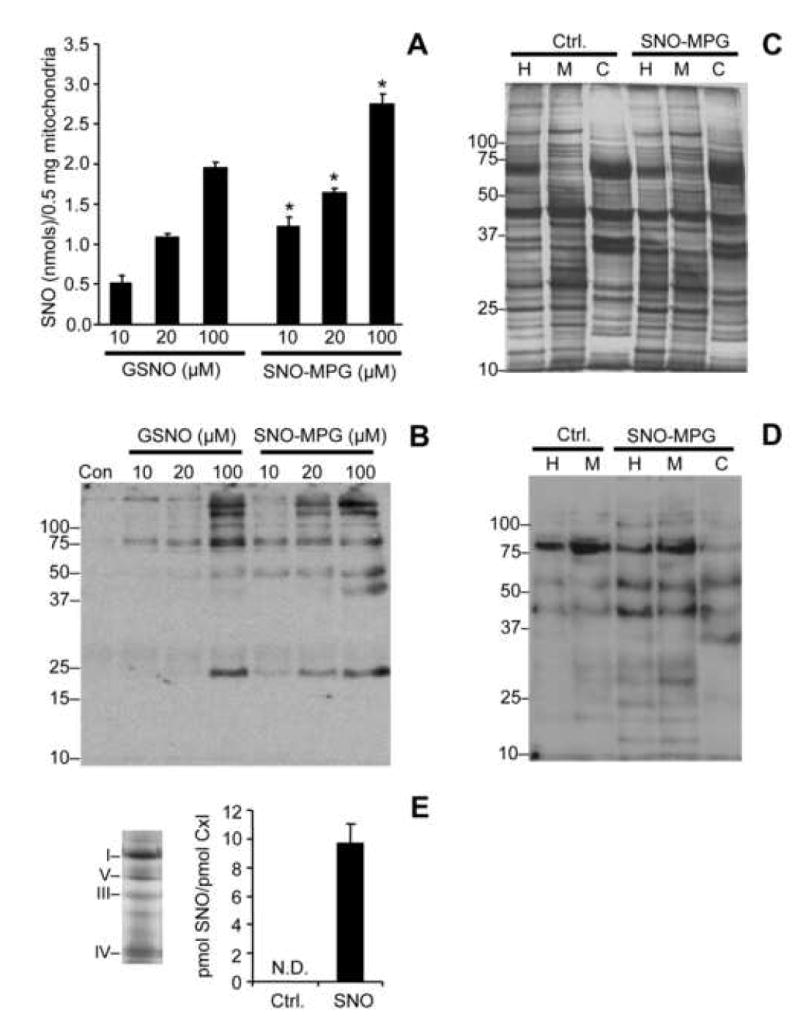
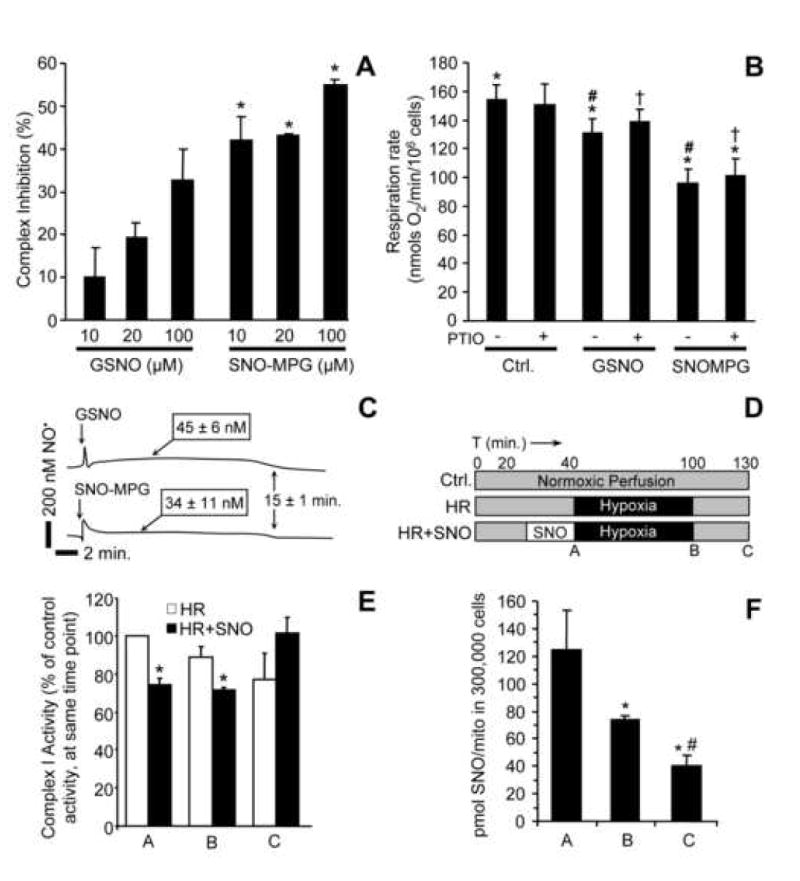
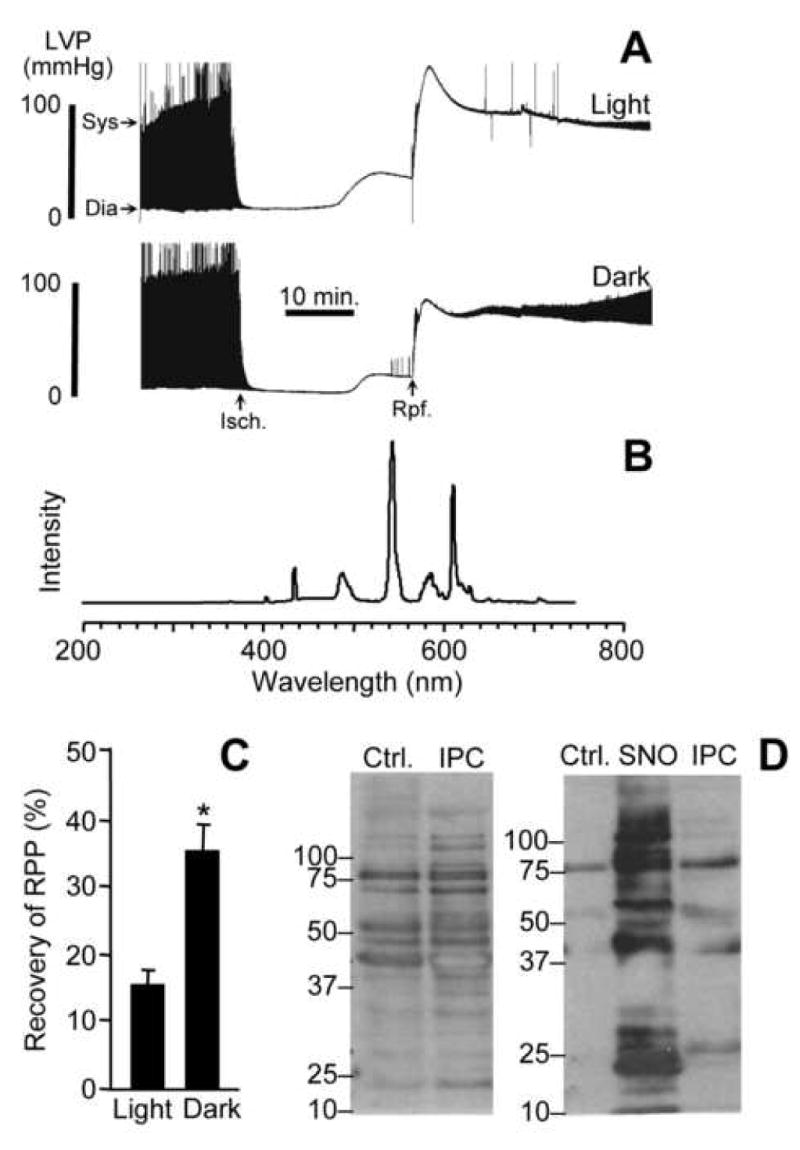
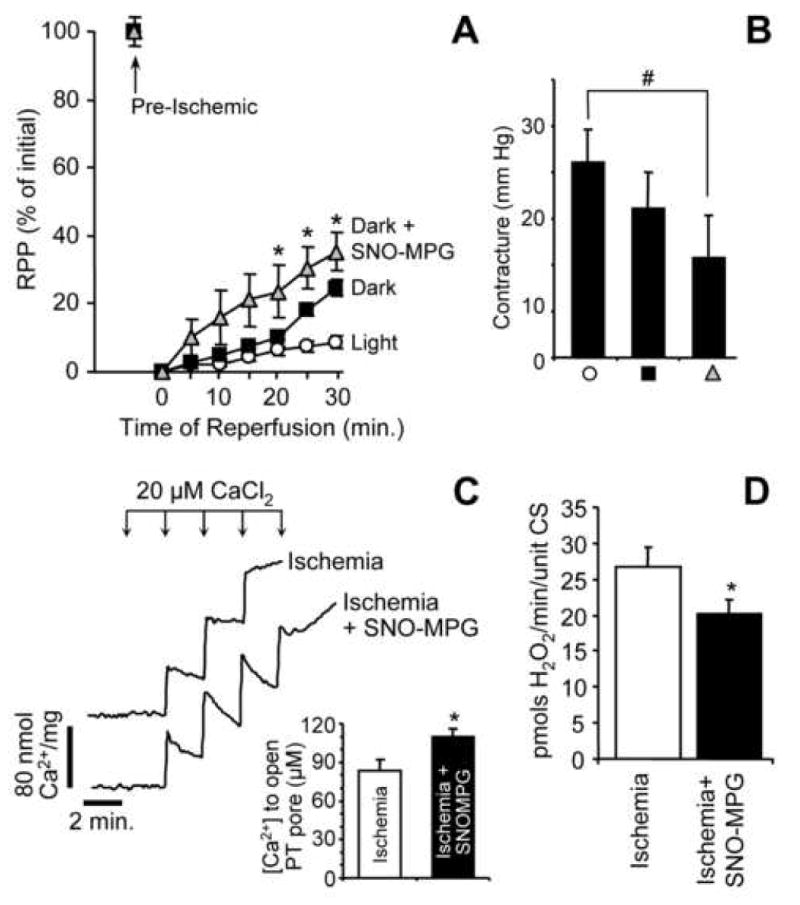
Mitochondrial dysfunction is a key pathologic event in cardiac ischemia-reperfusion (IR) injury, and protection of mitochondrial function is a potential mechanism underlying ischemic preconditioning (IPC). Acknowledging the role of nitric oxide (NO()) in IPC, it was hypothesized that mitochondrial protein S-nitrosation may be a cardioprotective mechanism. The reagent S-nitroso-2-mercaptopropionyl-glycine (SNO-MPG) was therefore developed to enhance mitochondrial S-nitrosation and elicit cardioprotection. Within cardiomyocytes, mitochondrial proteins were effectively S-nitrosated by SNO-MPG. Consistent with the recent discovery of mitochondrial complex I as an S-nitrosation target, SNO-MPG inhibited complex I activity and cardiomyocyte respiration. The latter effect was insensitive to the NO() scavenger c-PTIO, indicating no role for NO()-mediated complex IV inhibition. A cardioprotective role for reversible complex I inhibition has been proposed, and consistent with this SNO-MPG protected cardiomyocytes from simulated IR injury. Further supporting a cardioprotective role for endogenous mitochondrial S-nitrosothiols, patterns of protein S-nitrosation were similar in mitochondria isolated from Langendorff perfused hearts subjected to IPC, and mitochondria or cells treated with SNO-MPG. The functional recovery of perfused hearts from IR injury was also improved under conditions which stabilized endogenous S-nitrosothiols (i.e. dark), or by pre-ischemic administration of SNO-MPG. Mitochondria isolated from SNO-MPG-treated hearts at the end of ischemia exhibited improved Ca(2+) handling and lower ROS generation. Overall these data suggest that mitochondrial S-nitrosation and complex I inhibition constitute a protective signaling pathway that is amenable to pharmacologic augmentation.
Figures
References
-
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. - PubMed
-
- Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–41. - PubMed
-
- Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc Res. 2006;70:191–9. - PubMed
-
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
